Capítulo 6 Evolución temporal de un sistema de 2 estados — El máser de amoníaco y las oscilaciones cuánticas¶
Resumen de los capítulos anteriores:
En Cap. 4 aprendimos las 3 leyes de Feynman: "el cuadrado del valor absoluto de la amplitud es la probabilidad", "se suman las amplitudes de cada camino" y "se multiplican las amplitudes de procesos sucesivos". En Cap. 5, a través del experimento de Stern-Gerlach, vimos que el estado de una partícula de espín 1/2 puede escribirse como superposición de dos estados base, y que sus coeficientes (amplitudes de probabilidad) se comportan como componentes de un vector de números complejos. El "esqueleto" del espacio de estados está listo — pero aún no hemos introducido el tiempo.
Objetivo de este capítulo
- Establecer la ecuación diferencial que describe cómo cambian con el tiempo las amplitudes de probabilidad de un sistema de 2 estados, y al obtener los autovalores y autovectores de la matriz hamiltoniana, derivar los estados estacionarios y las oscilaciones cuánticas (oscilaciones de Rabi)
- Como ejemplo concreto, tratar el movimiento de inversión de la molécula de amoníaco NH₃ y el principio de funcionamiento del máser de amoníaco, experimentando "por qué el hamiltoniano está en el centro de la mecánica cuántica"
6.1 Ecuación fundamental de la evolución temporal — ¿Con qué regla cambian las amplitudes en el tiempo?¶
🟡 Lina: En los capítulos anteriores, aprendimos que el estado de un sistema de 2 estados se puede escribir como
\(C_1 = \langle 1|\psi\rangle\), \(C_2 = \langle 2|\psi\rangle\) son amplitudes de probabilidad complejas que satisfacen \(|C_1|^2 + |C_2|^2 = 1\).
🔵 Kai: Sí. Pero esto es una "foto instantánea", ¿verdad? Cuando pasa el tiempo, ¿cómo cambian \(C_1\) y \(C_2\)?
🟡 Lina: Buena pregunta. Ese es precisamente el tema de este capítulo. En mecánica clásica, la ecuación de movimiento de Newton \(F = ma\) determinaba cómo cambia la posición de un objeto en el tiempo. En mecánica cuántica también existe una ecuación que determina cómo cambian las amplitudes en el tiempo.
🔵 Kai: ¿Es como la "ecuación de movimiento" versión mecánica cuántica?
🟡 Lina: Así es. Aquí la aceptaremos como algo dado — es decir, como una "hipótesis fundamental consistente con los hechos experimentales". Pero esta forma tiene razones. Los requisitos son tres.
Primero, la ecuación debe ser lineal. Esto es para preservar el principio de superposición.
Segundo, la probabilidad debe conservarse. \(|C_1|^2 + |C_2|^2 = 1\) debe cumplirse siempre.
Tercero, la ecuación debe ser de primer orden.
🔵 Kai: ¿Por qué de primer orden? La ecuación de movimiento de Newton era de segundo orden, ¿no?
🟡 Lina: Buena comparación. Como la ecuación de Newton es de segundo orden, era necesario especificar tanto la posición como la velocidad como condiciones iniciales. Pero en mecánica cuántica, el vector de estado \(|\psi\rangle\) — es decir, el conjunto de amplitudes \(C_1(t), C_2(t)\) — contiene toda la información del sistema. En el caso de la molécula de amoníaco, si conoces los 2 números complejos \(C_1(t)\) y \(C_2(t)\), puedes determinar todo: la "probabilidad de que el nitrógeno esté arriba", la "probabilidad de que esté abajo", e incluso cómo cambiarán en el futuro.
🔵 Kai: Mmm, lo de "contiene toda la información" implica "basta con primer orden", pero todavía no me queda del todo claro...
🟡 Lina: Piénsalo así. En mecánica clásica, conocer solo la posición \(x\) de una pelota no te permite saber su movimiento futuro, ¿verdad? — sin conocer también la velocidad \(v\), no puedes distinguir si está quieta o moviéndose. Por eso la ecuación de Newton es de segundo orden, y se necesitan tanto \(x\) como \(v\) como condiciones iniciales.
Pero en mecánica cuántica, el conjunto de amplitudes \(C_1, C_2\) es la descripción completa del sistema — es decir, toda la información correspondiente a la "posición" y la "velocidad" está contenida aquí. Si la ecuación fuera de segundo orden, además de \(C_1(0)\), \(C_2(0)\) necesitarías especificar \(dC_1/dt|_0\), \(dC_2/dt|_0\) para que la solución fuera única. Eso significaría que "las amplitudes solas no son información suficiente", lo cual contradiría la hipótesis de que "las amplitudes son una descripción completa". Por eso la ecuación debe ser de primer orden.
Es decir, si aceptas la hipótesis de que "las amplitudes son la descripción completa del sistema", que la ecuación sea de primer orden es una consecuencia lógica. Y la hipótesis misma la aceptamos como un principio fundamental consistente con los hechos experimentales.
🔵 Kai: Que "las amplitudes por sí solas sean completas" es algo que por ahora solo podemos aceptar como dado, ¿no?
🟡 Lina: Así es. Si esta hipótesis es correcta o no se juzga por si las predicciones derivadas de ella coinciden con los experimentos — precisamente en este capítulo predeciremos la frecuencia de oscilación de la molécula de amoníaco y la compararemos con el experimento como verificación. Bien, la ecuación diferencial lineal más general que satisface estos 3 requisitos tiene la siguiente forma:
La razón por la que no hay términos constantes (términos que no son proporcionales a \(C_1\) o \(C_2\)) en el lado derecho es que, si los hubiera, incluso con \(C_1 = C_2 = 0\) (estado de vacío) surgirían amplitudes espontáneamente — esto significaría que "la probabilidad surge de la nada", lo cual es físicamente absurdo.
🔵 Kai: Vaya, ecuaciones diferenciales de entrada... \(\hbar\) es la constante de Planck reducida que apareció antes, ¿verdad? La \(i\) del lado izquierdo es la unidad imaginaria. Pero, ¿qué son \(H_{11}\) y \(H_{12}\) del lado derecho? ¿Qué significan los subíndices?
🟡 Lina: Esos son los elementos de la matriz hamiltoniana. El significado de los subíndices es: el primer subíndice \(i\) de \(H_{ij}\) representa la "fila" (es decir, de qué ecuación de amplitud se trata en el lado izquierdo), y el segundo subíndice \(j\) representa la "columna" (es decir, a qué amplitud multiplica en el lado derecho). Por ejemplo, \(H_{12}\) es "el coeficiente de \(C_2\) (columna 2) que aparece en la ecuación de \(C_1\) (fila 1)". Son cantidades con dimensiones de energía y, en general, son números complejos. Explicaré su significado físico en detalle en la siguiente sección, pero por ahora piensa en ellas como "constantes relacionadas con la energía". Primero quiero que aprecies la "forma" de la ecuación.
⚪ Mei: El lado izquierdo es "la tasa de cambio temporal de \(C_1\)" multiplicada por \(i\hbar\). El lado derecho es una combinación lineal de \(C_1\) y \(C_2\). Es decir, el cambio de una amplitud depende de los valores de ambas amplitudes.
🟡 Lina: Exacto. \(C_1\) y \(C_2\) evolucionan en el tiempo "influyéndose mutuamente" — esta es la ecuación de movimiento del sistema de 2 estados en mecánica cuántica.
🔵 Kai: ¿Cómo quedarían las ecuaciones (6.2a) y (6.2b) escritas en forma matricial?
🟡 Lina: Buen instinto. Se pueden escribir así:
La matriz \(2 \times 2\) del lado derecho es la matriz hamiltoniana \(H\).
✅ Verificación de comprensión: En la ecuación (6.3), \(i\hbar\) multiplicando el lado izquierdo es una constante. Si \(H_{12} = H_{21} = 0\), ¿qué sucedería con \(C_1\) y \(C_2\)? (Pista: el sistema de ecuaciones se "desacopla".)
Respuesta
Si \(H_{12} = H_{21} = 0\), la ecuación (6.2a) se convierte en \(i\hbar\,dC_1/dt = H_{11}\,C_1\), sin depender de \(C_2\). Igualmente, la ecuación (6.2b) se convierte en \(i\hbar\,dC_2/dt = H_{22}\,C_2\). Es decir, \(C_1\) y \(C_2\) evolucionan independientemente en el tiempo. Los elementos no diagonales \(H_{12}\), \(H_{21}\) desempeñan el papel de "conectar los dos estados".
6.2 Introducción de la matriz hamiltoniana — Una matriz que representa la energía¶
🔵 Kai: La matriz hamiltoniana, ¿qué representa al fin y al cabo?
🟡 Lina: En una palabra, es una matriz que contiene toda la información sobre la energía del sistema. Su nombre proviene de Hamilton, quien en mecánica clásica llamó hamiltoniano a la función que representa la energía total del sistema (energía cinética + energía potencial).
🔵 Kai: ¿Entonces es la "versión cuántica" de la energía total clásica?
🟡 Lina: Puedes pensarlo así. Sin embargo, en mecánica cuántica, la energía no es "un solo número" sino una "matriz". La razón es que, como hay 2 estados, no basta con describir "la energía del estado \(|1\rangle\)" y "la energía del estado \(|2\rangle\)" — también hay que describir "el acoplamiento entre los estados \(|1\rangle\) y \(|2\rangle\)".
⚪ Mei: Ya veo, un solo número no es suficiente, y como se necesitan tantas "casillas" como estados haya, se convierte en una matriz.
🟡 Lina: Así es. Organizando el significado de cada componente:
Tabla 6.1: Significado físico de cada componente de la matriz hamiltoniana
| Componente | Significado |
|---|---|
| \(H_{11}\) | "Energía propia" estando en el estado \(\|1\rangle\) |
| \(H_{22}\) | "Energía propia" estando en el estado \(\|2\rangle\) |
| \(H_{12}\) | "Intensidad de la amplitud de transición" del estado \(\|2\rangle\) al estado \(\|1\rangle\) (el primer subíndice es el "destino", el segundo es el "origen") |
| \(H_{21}\) | "Intensidad de la amplitud de transición" del estado \(\|1\rangle\) al estado \(\|2\rangle\) (igualmente, 2 es destino, 1 es origen) |
🔵 Kai: Los elementos diagonales son energía, y los no diagonales son la "conexión" entre estados, ¿no?
🟡 Lina: Así es. Y la matriz hamiltoniana tiene una propiedad importante. La hermiticidad (propiedad hermítica):
Esto significa que "la matriz que se obtiene al tomar el complejo conjugado de cada componente y luego intercambiar filas y columnas es igual a la matriz original". Concretamente, el complejo conjugado de \(H_{12}\) es igual a \(H_{21}\). Para los elementos diagonales \(H_{11}\), \(H_{22}\), como \(H_{11}^* = H_{11}\), \(H_{22}^* = H_{22}\), los elementos diagonales son reales.
🔵 Kai: ¿Por qué tiene que ser hermítica?
🟡 Lina: La razón es la conservación de la probabilidad. Para que \(|C_1|^2 + |C_2|^2 = 1\) se cumpla siempre, la matriz hamiltoniana debe ser hermítica. Veámoslo brevemente. Si exigimos \(d(|C_1|^2 + |C_2|^2)/dt = 0\) y calculamos el lado izquierdo usando la ecuación (6.2), obtenemos la condición \(H_{12} = H_{21}^*\) — que es precisamente la hermiticidad.
🔵 Kai: Lo del complejo conjugado me preocupa un poco...
🟡 Lina: Te daré solo una pista del cálculo. Como \(|C_1|^2 = C_1^* C_1\), al derivar usamos la regla del producto: \(d|C_1|^2/dt = (dC_1^*/dt)C_1 + C_1^*(dC_1/dt)\). \(dC_1/dt\) se obtiene de la ecuación (6.2a) como \(dC_1/dt = (H_{11}C_1 + H_{12}C_2)/(i\hbar)\). \(dC_1^*/dt\) se obtiene tomando el complejo conjugado de ambos lados de la ecuación (6.2a) — al tomar el complejo conjugado, \(i\) se convierte en \(-i\), así que el lado izquierdo se convierte en \(-i\hbar\,dC_1^*/dt\), y el lado derecho en \(H_{11}^*\,C_1^* + H_{12}^*\,C_2^*\). Sustituyendo y simplificando, se obtiene \(H_{12} = H_{21}^*\). La demostración detallada la puedes verificar en el ejercicio (Problema B-1. Cálculo del factor de fase de un estado estacionario).
📝 Ejercicios:
- Derivar la hermiticidad \(H_{12} = H_{21}^*\) a partir de \(d(|C_1|^2 + |C_2|^2)/dt = 0\) y la ecuación (6.2). → Problema B-1. Cálculo del factor de fase de un estado estacionario
Intuitivamente, corresponde a que "la energía debe ser real".
⚪ Mei: Es decir, el requisito físico de que "la energía debe ser real" se expresa como una propiedad matemática de la matriz (hermiticidad).
🟡 Lina: Bien observado. La discusión sobre autovalores vendrá enseguida.
Por qué \(i\hbar\) está en el lado izquierdo¶
🔵 Kai: Una cosa que me preguntaba: el \(i\hbar\) del lado izquierdo de la ecuación (6.2), ¿para qué está? ¿No bastaría con \(dC_1/dt = (\text{algo}) \times C_1 + \cdots\)?
🟡 Lina: Buena duda. Si no hubiera \(i\), es decir, si fuera
entonces, cuando \(H_{11}\) es real, \(C_1\) crecería o decaería exponencialmente permaneciendo real. Esto no conservaría la probabilidad. Con la \(i\), la solución se vuelve oscilatoria (la fase rota), y \(|C_1|^2\) se mantiene constante.
🔵 Kai: Ya veo. La \(i\) tiene el papel de generar "rotación" en vez de "crecimiento/decaimiento".
🟡 Lina: Así es. Y \(\hbar\) está para ajustar las unidades. \(C\) es una amplitud de probabilidad adimensional, así que \(dC/dt\) tiene dimensiones de \([\text{tiempo}]^{-1}\). Las dimensiones de \(\hbar\) son \([\text{energía} \times \text{tiempo}]\), así que el lado izquierdo \(i\hbar\,dC/dt\) tiene dimensiones de \([\text{energía} \times \text{tiempo}] \times [\text{tiempo}]^{-1} = [\text{energía}]\). El lado derecho \(H_{ij} C_j\) también tiene dimensiones de energía porque \(H_{ij}\) tiene dimensiones de energía, así que todo cuadra.
🔵 Kai: Entiendo. Entonces resumiendo: la \(i\) es "para conservar la probabilidad (para generar oscilaciones)", \(\hbar\) es "para ajustar las dimensiones", y que el lado derecho sea una combinación lineal de \(C_1\) y \(C_2\) es "para preservar el principio de superposición" — los 3 requisitos corresponden a cada parte de la ecuación.
🟡 Lina: Exactamente. Bien resumido.
✅ Verificación de comprensión: La hermiticidad de la matriz hamiltoniana \(H_{ij}^* = H_{ji}\), ¿de qué requisito físico proviene?
Respuesta
Proviene del requisito de que la probabilidad total \(|C_1|^2 + |C_2|^2 = 1\) no cambie en el tiempo. La hermiticidad también garantiza que los autovalores del hamiltoniano (energías) sean reales.
6.3 Modelo de 2 estados de la molécula de amoníaco¶
🟡 Lina: Bien, ahora pasemos a un ejemplo concreto. La molécula de amoníaco NH₃.
🔵 Kai: ¿El amoníaco, ese del olor penetrante...?
🟡 Lina: Sí. Es una molécula formada por 1 átomo de nitrógeno (N) y 3 átomos de hidrógeno (H), con forma piramidal. Los 3 átomos de hidrógeno forman un plano triangular, y el átomo de nitrógeno se sitúa un poco por encima de ese plano (Fig. 6.1「Estructura de la molécula de amoníaco」).
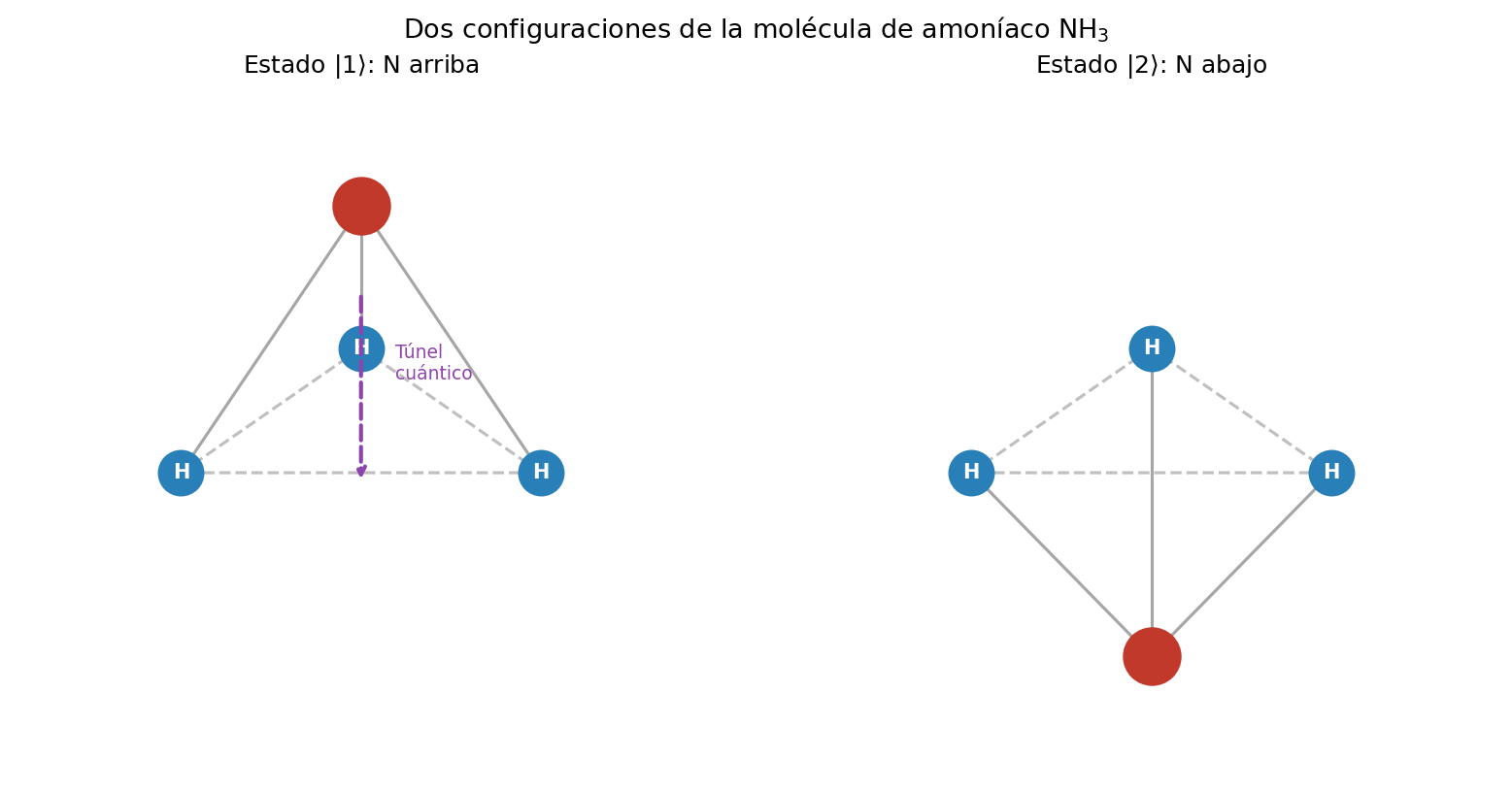
Fig. 6.1: Estructura de la molécula de amoníaco. Estructura de la molécula de amoníaco NH₃. El átomo de nitrógeno (N) se sitúa arriba o abajo del plano formado por los 3 átomos de hidrógeno (H).
🔵 Kai: Forma piramidal... eso significa que según si el nitrógeno está "arriba" o "abajo", ¡hay dos configuraciones!
🟡 Lina: Exacto. Aquí hacemos una aproximación audaz. Fijamos la rotación, vibración y traslación de la molécula, y el único grado de libertad que queda es de qué lado del plano de hidrógenos está el átomo de nitrógeno. Con esto tenemos un sistema de 2 estados.
- Estado \(|1\rangle\): Configuración con el nitrógeno "arriba" del plano
- Estado \(|2\rangle\): Configuración con el nitrógeno "abajo" del plano
⚪ Mei: En la molécula real hay muchos más grados de libertad, pero los "congelamos" y nos enfocamos solo en el arriba/abajo del nitrógeno — esa es la aproximación.
🟡 Lina: Así es. Esta aproximación se justifica cuando las escalas de energía de los otros grados de libertad son mucho mayores que la escala de energía del movimiento de inversión del nitrógeno. De hecho, la energía de excitación electrónica es de varios eV, las vibraciones moleculares son del orden de 0.1 eV, mientras que la energía asociada al movimiento de inversión del nitrógeno es del orden de \(10^{-4}\) eV — órdenes de magnitud menor.
Determinación del hamiltoniano¶
🟡 Lina: Ahora determinemos la matriz hamiltoniana de este sistema de 2 estados. La pista es la simetría.
🔵 Kai: ¿Simetría?
🟡 Lina: Mira de nuevo Fig. 6.1「Estructura de la molécula de amoníaco」. Considera la operación de voltear la molécula de amoníaco arriba-abajo. La configuración con el nitrógeno "arriba" se intercambia exactamente con la configuración del nitrógeno "abajo". Es decir, los estados \(|1\rangle\) y \(|2\rangle\) son físicamente completamente equivalentes.
⚪ Mei: Eso significa que \(H_{11} = H_{22}\). La "energía propia" de ambas configuraciones es la misma.
🟡 Lina: Así es. Escribiremos este valor común como \(E_0\):
✅ Verificación de comprensión: En el hamiltoniano de la molécula de amoníaco, ¿por qué razón física se tiene \(H_{11} = H_{22}\)?
Respuesta
Al invertir arriba-abajo la molécula de amoníaco, la configuración con nitrógeno "arriba" y la de "abajo" se intercambian. Como la molécula es físicamente equivalente bajo esta operación, la "energía propia" de ambas configuraciones debe tener el mismo valor \(E_0\).
Ahora los elementos no diagonales. Que el átomo de nitrógeno "atraviese" el plano de hidrógenos y pase al otro lado — en mecánica clásica esto sería imposible si no hay suficiente energía, ¿verdad? Pero en mecánica cuántica ocurre el efecto túnel cuántico (quantum tunneling). En mecánica clásica, si la energía es menor que la altura de la barrera, el paso es absolutamente imposible, pero en mecánica cuántica la amplitud de probabilidad de "filtrarse" al otro lado de la barrera no es cero.
🔵 Kai: ¡Efecto túnel! En Cap. 3 con la doble rendija también se habló de "caminos clásicamente imposibles".
🟡 Lina: Buena conexión. Escribiremos esta amplitud de "filtrarse" como \(-A\) (con \(A > 0\)). El signo negativo es convencional y hace que los cálculos posteriores sean más claros. Por simetría \(H_{12} = H_{21}\), así que:
⚪ Mei: La hermiticidad \(H_{12}^* = H_{21}\) también se satisface. Si \(A\) es real, entonces \((-A)^* = -A = H_{21}\).
🟡 Lina: En resumen, la matriz hamiltoniana de la molécula de amoníaco es:
🔵 Kai: Es simple. Pero, ¿cómo se determina el valor de \(A\)?
🟡 Lina: \(A\) está relacionada con la amplitud de probabilidad de que el átomo de nitrógeno atraviese por efecto túnel la barrera de potencial. En principio puede calcularse a partir de la altura y anchura de la barrera, pero en la práctica se usa el valor medido experimentalmente. Para la molécula de amoníaco, se sabe que la frecuencia correspondiente a \(2A\) es aproximadamente 24,000 MHz (microondas con longitud de onda de aproximadamente 1.25 cm).
🔵 Kai: Es la región de microondas.
📝 Ejercicios:
- Cuando \(2A = hf\) con \(f = 24{,}000\) MHz, hallar \(A\) en unidades de eV. → Problema B-2. Cálculo de la energía de desdoblamiento por efecto túnel
6.4 Autovalores y autovectores — Encontrando los estados estacionarios¶
🟡 Lina: Ya que hemos determinado la matriz hamiltoniana (6.7), ahora resolvamos la ecuación (6.2). Primero busquemos los estados estacionarios — estados especiales cuyo "carácter" no cambia con el tiempo.
🔵 Kai: ¿Qué significa que "el carácter del estado no cambie"?
🟡 Lina: Es un estado donde la razón \(C_1 : C_2\) de las amplitudes permanece constante en el tiempo. Como la ecuación (6.2) tiene la forma "la derivada es proporcional a la función original", es natural probar funciones exponenciales como solución — la misma idea que cuando aprendiste que la solución de \(dy/dx = ky\) es \(e^{kx}\). Concretamente:
donde \(a_1\), \(a_2\) son constantes independientes del tiempo, y \(E\) es una constante (con dimensiones de energía).
⚪ Mei: Como ambas amplitudes oscilan con la misma frecuencia \(E/\hbar\), la razón \(C_1/C_2 = a_1/a_2\) no depende del tiempo.
🟡 Lina: Así es. Sustituyamos (6.8) en (6.2). El lado izquierdo es:
El lado derecho es:
Dividiendo ambos lados por \(e^{-iEt/\hbar}\):
Igualmente, de la ecuación (6.2b):
🔵 Kai: ¡Ah, la ecuación diferencial desapareció y quedó un simple sistema de ecuaciones algebraicas!
🟡 Lina: Así es. Escrito en forma matricial:
🟡 Lina: Esto es lo que en álgebra lineal se llama un problema de autovalores (eigenvalue problem). Es el problema de encontrar un vector especial \(\begin{pmatrix} a_1 \\ a_2 \end{pmatrix}\) y un escalar \(E\) tales que al multiplicar la matriz \(H\) por el vector, se obtiene un múltiplo escalar \(E\) del mismo vector. \(E\) se llama autovalor (eigenvalue) y \(\begin{pmatrix} a_1 \\ a_2 \end{pmatrix}\) se llama autovector (eigenvector). "Eigen" es una palabra alemana que significa "propio, característico" — es decir, "valores y vectores especiales que solo pertenecen a esa matriz".
🔵 Kai: ¿Por qué es especial un vector que "se convierte en un múltiplo escalar de sí mismo"? ¿En qué se diferencia de un sistema de ecuaciones normal?
🟡 Lina: Te respondo desde dos perspectivas. Primero, la diferencia con un sistema de ecuaciones normal. Los sistemas de ecuaciones que resolviste en la preparatoria eran del tipo "\(2x + 3y = 5\), \(x - y = 1\)", donde el lado derecho son constantes fijas y buscas un conjunto de valores para \(x, y\). Pero en el problema de autovalores, "el lado derecho es un múltiplo escalar de las propias incógnitas" — es decir, el escalar \(E\) mismo también es desconocido, y buscas \(E\) y el vector simultáneamente. Además, generalmente hay múltiples conjuntos de soluciones — en este caso, 2 conjuntos.
🔵 Kai: Ya veo, \(E\) también es parte de las incógnitas... Entonces hay más incógnitas que de costumbre.
🟡 Lina: Ahora la imagen geométrica. En general, al multiplicar un vector por una matriz, tanto la dirección como la magnitud del vector cambian. Pero los autovectores son especiales: al multiplicarlos por la matriz, la dirección no cambia — solo cambia la magnitud (el múltiplo escalar). Por ejemplo, \(\begin{pmatrix} 2 & 1 \\ 0 & 3 \end{pmatrix}\begin{pmatrix} 1 \\ 0 \end{pmatrix} = \begin{pmatrix} 2 \\ 0 \end{pmatrix} = 2\begin{pmatrix} 1 \\ 0 \end{pmatrix}\) — el vector \(\begin{pmatrix} 1 \\ 0 \end{pmatrix}\) no cambia de dirección al multiplicarlo por la matriz, solo se duplica. Es decir, encontrar "direcciones estables bajo la acción de la matriz" es el problema de autovalores.
⚪ Mei: "La dirección no cambia bajo la acción de la matriz = el carácter no cambia con el tiempo" — la correspondencia entre matemáticas y física es elegante.
🟡 Lina: Y el significado físico. Recuerda. Estábamos buscando estados donde la razón de amplitudes \(C_1 : C_2\) fuera constante en el tiempo, ¿verdad? Asumir el factor común \(e^{-iEt/\hbar}\) en (6.8) era precisamente para eso. Si encontramos la solución del problema de autovalores, corresponde a un estado estacionario — es decir, un estado con energía definida. El autovalor \(E\) es la energía de ese estado estacionario.
Cálculo de los autovalores¶
🟡 Lina: Reescribamos la ecuación (6.10). Primero introduzco una notación. Llamamos matriz identidad (identity matrix) a \(\mathbb{1} = \begin{pmatrix} 1 & 0 \\ 0 & 1 \end{pmatrix}\). Es la matriz con 1 en la diagonal y 0 en las demás posiciones. Si la multiplicas por un vector: \(\begin{pmatrix} 1 & 0 \\ 0 & 1 \end{pmatrix}\begin{pmatrix} a_1 \\ a_2 \end{pmatrix} = \begin{pmatrix} a_1 \\ a_2 \end{pmatrix}\) — devuelve el vector original. Es decir, la matriz identidad es el "1" del mundo matricial — así como \(1 \times x = x\) en el mundo de los números, \(\mathbb{1} \times \mathbf{a} = \mathbf{a}\).
🔵 Kai: Es la versión matricial de "la operación que no hace nada".
🟡 Lina: Así es. Denotando \(\mathbf{a} = \begin{pmatrix} a_1 \\ a_2 \end{pmatrix}\), el lado derecho \(E\mathbf{a}\) es lo mismo que \(E\mathbb{1}\mathbf{a}\). La razón es que \(E\mathbb{1} = \begin{pmatrix} E & 0 \\ 0 & E \end{pmatrix}\), y al multiplicar esto por el vector se obtiene \(\begin{pmatrix} E\,a_1 \\ E\,a_2 \end{pmatrix} = E\begin{pmatrix} a_1 \\ a_2 \end{pmatrix}\) — es decir, el mismo resultado que multiplicar cada componente por \(E\). Pasando al lado izquierdo queda \((H - E\mathbb{1})\mathbf{a} = 0\). En componentes: \(\begin{pmatrix} E_0 - E & -A \\ -A & E_0 - E \end{pmatrix}\begin{pmatrix} a_1 \\ a_2 \end{pmatrix} = \begin{pmatrix} 0 \\ 0 \end{pmatrix}\) — simplemente se resta \(E\) de los elementos diagonales. Es el problema de encontrar un vector no nulo \(\mathbf{a}\) tal que "al multiplicar por la matriz \((H - E\mathbb{1})\) se obtiene el vector cero".
🔵 Kai: Con \(\mathbf{a} = 0\) se cumple trivialmente, pero eso no tiene sentido, ¿verdad? ¿Cuál es la condición para que existan soluciones no nulas?
🟡 Lina: Lo que queremos hacer es encontrar "la condición para que \((H - E\mathbb{1})\mathbf{a} = 0\) tenga solución con \(\mathbf{a} \neq 0\)". En componentes, esto es un sistema \(ax + by = 0\), \(cx + dy = 0\) (donde \(a, b, c, d\) son los elementos de \((H - E\mathbb{1})\)) y buscamos soluciones con \(x \neq 0\). De la primera ecuación \(x = -by/a\) (asumiendo \(a \neq 0\)), sustituyendo en la segunda: \((ad - bc)y/a = 0\). Para que exista solución con \(y \neq 0\) se necesita \(ad - bc = 0\).
🔵 Kai: ¿Y si \(a = 0\)?
🟡 Lina: Buena pregunta. Si seguimos todos los casos se alarga, pero la conclusión es que, incluso cuando \(a = 0\), la condición "existen soluciones no triviales ⟺ \(ad - bc = 0\)" no cambia. Por ejemplo, si \(a = 0\) y \(b \neq 0\), la primera ecuación es \(by = 0\), de donde solo \(y = 0\) — en este caso \(ad - bc = -bc \neq 0\), así que "determinante no nulo → no hay soluciones no triviales" es consistente. Examines el caso que examines, la conclusión es la misma — la condición para soluciones no nulas es siempre \(ad - bc = 0\).
🔵 Kai: Entiendo, las dos ecuaciones dicen esencialmente "lo mismo" — solo hay 1 ecuación independiente de información, ¿no?
🟡 Lina: ¡Exacto! Esta cantidad \(ad - bc\) se llama el determinante de la matriz \(2 \times 2\) \(\begin{pmatrix} a & b \\ c & d \end{pmatrix}\), y se denota con el símbolo \(\det\):
Intuitivamente, el determinante mide "cuán independientes son las 2 ecuaciones" — si es cero, las 2 ecuaciones solo contienen la información de 1; si no es cero, las 2 son independientes y la única solución es \(x = y = 0\).
🔵 Kai: Cuando el determinante no es cero, solo queda \(x = y = 0\)... dicho al revés, solo cuando el determinante es cero existen soluciones con \(x \neq 0\).
⚪ Mei: Dicho de otro modo, que el determinante sea cero significa que "las 2 ecuaciones solo contienen la información de 1" — por eso solo se determina la razón de las incógnitas, y la escala total queda libre. Eso es lo que significa "existen soluciones no nulas".
🟡 Lina: Exacto. Como complemento, cuando el determinante no es cero, la matriz tiene una matriz inversa (inverse matrix). La matriz inversa es la versión matricial del recíproco de un número \(a^{-1}\) (\(a \times a^{-1} = 1\)): una matriz \(M^{-1}\) que satisface \(M M^{-1} = \mathbb{1}\) (matriz identidad). Si existiera la inversa, al multiplicar ambos lados de \((H - E\mathbb{1})\mathbf{a} = 0\) por la izquierda con \(M^{-1}\), obtendríamos \(\mathbf{a} = M^{-1} \cdot 0 = 0\) — es decir, "determinante no nulo → la única solución es \(\mathbf{a} = 0\)". El contrarrecíproco es "\(\mathbf{a} \neq 0\) es solución → determinante es cero". Ahora basta con usar la conclusión "determinante cero ⟺ existen soluciones no nulas". Por lo tanto, la condición para que existan soluciones no nulas es que el determinante sea cero:
Simplificando:
🔵 Kai: ¡Ah, \((E_0 - E)^2 - A^2 = 0\) tiene la forma \(x^2 - a^2 = (x-a)(x+a)\)! Factorizando: \((E_0 - E - A)(E_0 - E + A) = 0\), así que...
🟡 Lina: Así es. Si \(E_0 - E = +A\) entonces \(E = E_0 - A\); si \(E_0 - E = -A\) entonces \(E = E_0 + A\). Es decir, obtenemos 2 autovalores:
🔵 Kai: ¿Eh? \(E_I\) tiene la energía más alta. Que I sea el de arriba y II el de abajo es un poco confuso.
🟡 Lina: Sí. Es la numeración según el libro de Feynman, pero recuerda que \(E_I > E_{II}\).
⚪ Mei: Lo que originalmente era un solo nivel de energía \(E_0\) se ha dividido en \(A\) hacia arriba y \(A\) hacia abajo debido a la amplitud de túnel \(A\). La diferencia de energía es \(E_I - E_{II} = 2A\).
🟡 Lina: Esto es el desdoblamiento túnel (tunnel splitting). Precisamente porque existe la posibilidad de que el átomo de nitrógeno atraviese el plano, los niveles de energía se dividen en dos. Si \(A = 0\) (sin efecto túnel), \(E_I = E_{II} = E_0\) y los dos niveles se superponen — un estado degenerado (degeneracy). Este comportamiento lo resumiremos en una figura más adelante.
Cálculo de los autovectores¶
🟡 Lina: Ahora encontremos los autovectores correspondientes a cada autovalor.
Caso del autovalor \(E_I = E_0 + A\):
Sustituyendo \(E = E_0 + A\) en la ecuación (6.9a):
De la condición de normalización \(|a_1|^2 + |a_2|^2 = 1\), se obtiene \(|a_1| = |a_2| = 1/\sqrt{2}\). Como \(a_1 = -a_2\), podemos elegir por ejemplo \(a_1 = 1/\sqrt{2}\), \(a_2 = -1/\sqrt{2}\) (multiplicar todo por \(e^{i\theta}\) representa el mismo estado físico, ya que como aprendimos en Cap. 5 hay libertad en la elección de la fase — aquí elegimos la opción real más simple). Por lo tanto:
Caso del autovalor \(E_{II} = E_0 - A\):
Sustituyendo \(E = E_0 - A\) en la ecuación (6.9a):
De la condición de normalización \(|a_1| = |a_2| = 1/\sqrt{2}\). Por lo tanto:
🔵 Kai: ¡Interesante! El estado de menor energía \(|II\rangle\) es la "suma" de \(|1\rangle\) y \(|2\rangle\), y el de mayor energía \(|I\rangle\) es la "resta".
🟡 Lina: Así es. \(|II\rangle\) tiene las amplitudes de nitrógeno arriba y abajo con el mismo signo — estado simétrico. \(|I\rangle\) tiene signos opuestos — estado antisimétrico. El estado simétrico tiene menor energía. Este es un patrón que aparecerá repetidamente cuando tratemos el pozo de potencial a partir de Cap. 9.
✅ Verificación de comprensión: De los autoestados \(|I\rangle\) y \(|II\rangle\), ¿cuál tiene menor energía? ¿Y es una superposición de \(|1\rangle\) y \(|2\rangle\) de qué tipo?
Respuesta
El de menor energía es \(|II\rangle = \frac{1}{\sqrt{2}}(|1\rangle + |2\rangle)\), con energía \(E_{II} = E_0 - A\). Es el estado simétrico que suma \(|1\rangle\) y \(|2\rangle\) con el mismo signo. El estado antisimétrico \(|I\rangle\) tiene mayor energía.
🟡 Lina: Verifiquemos que los dos autovectores son ortogonales. \(\langle I|II\rangle = \frac{1}{2}(\langle 1| - \langle 2|)(|1\rangle + |2\rangle) = \frac{1}{2}(1 + 0 - 0 - 1) = 0\). Son ortogonales.
⚪ Mei: Verificarlo explícitamente con el cálculo es muy satisfactorio. ¿Es una coincidencia, o se cumple en general?
🟡 Lina: Buena pregunta. En realidad no es coincidencia. Los autovectores de una matriz hermítica correspondientes a autovalores distintos siempre son ortogonales — es un teorema general del álgebra lineal. Físicamente, \(|I\rangle\) y \(|II\rangle\) son "estados completamente distinguibles". En Fig. 6.2「Relación entre la base de posición y la base de energía」, veamos geométricamente la relación entre las dos bases.
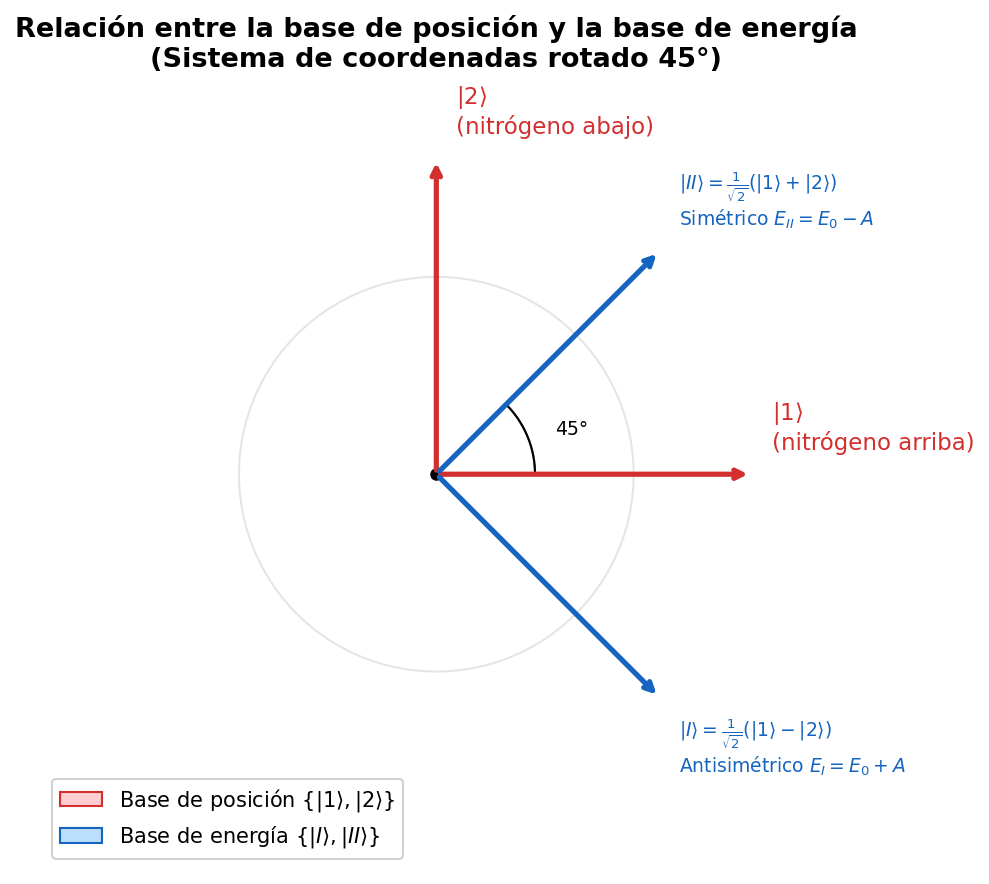
Fig. 6.2: Relación entre la base de posición y la base de energía. La "base de posición" \(\{|1\rangle, |2\rangle\}\) y la "base de energía" \(\{|I\rangle, |II\rangle\}\) están rotadas 45° entre sí. \(|II\rangle\) (simétrico) es la suma igual de \(|1\rangle\) y \(|2\rangle\); \(|I\rangle\) (antisimétrico) es la resta.
🟡 Lina: Resumamos los resultados hasta aquí en una figura. Si dibujamos la energía potencial de la molécula de amoníaco como función de la posición del nitrógeno, obtenemos dos valles (pozos) correspondientes a las dos posiciones estables "arriba" y "abajo", separados por una barrera central — esto se llama potencial de doble pozo (double-well potential) (Fig. 6.3「Potencial de doble pozo y desdoblamiento de energía」).
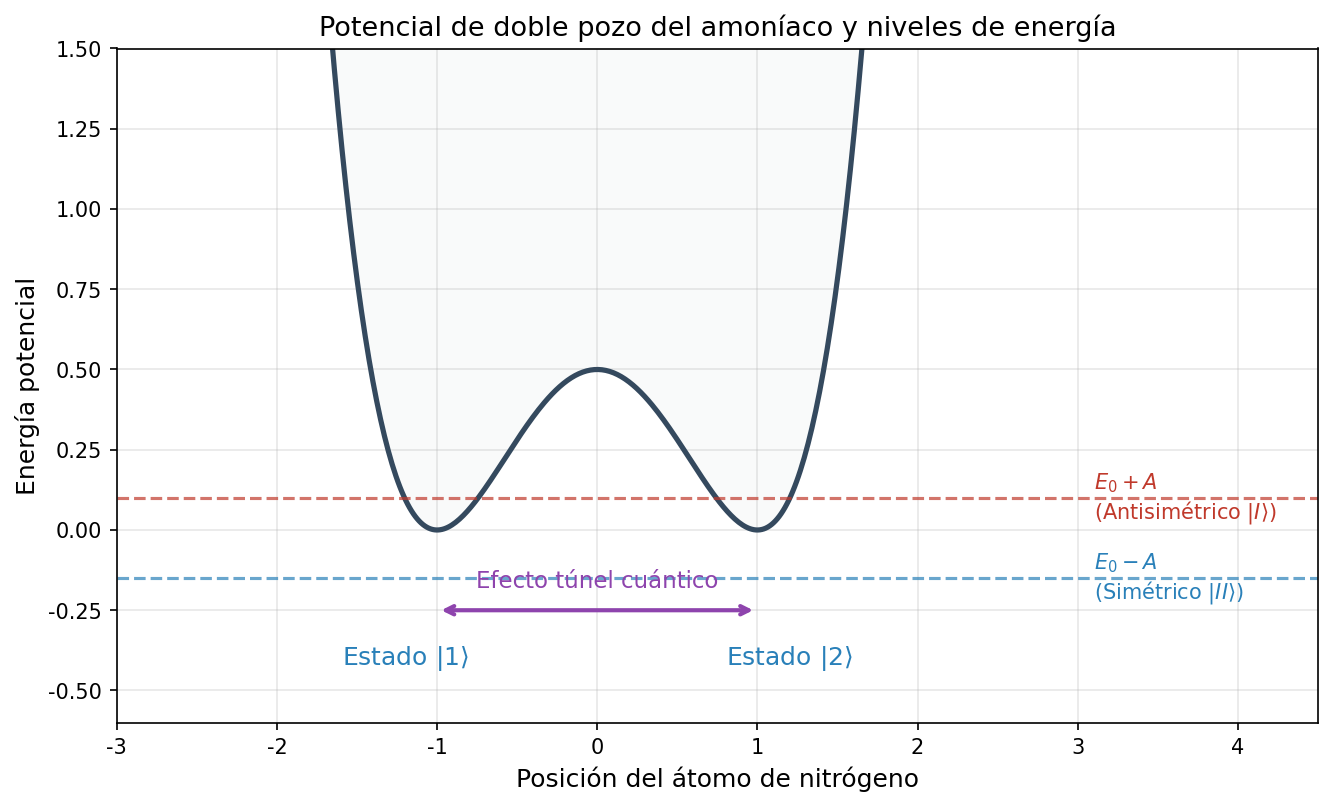
Fig. 6.3: Potencial de doble pozo y desdoblamiento de energía. Potencial de doble pozo de la molécula de amoníaco. Hay dos pozos correspondientes al estado \(|1\rangle\) (nitrógeno arriba) y al estado \(|2\rangle\) (nitrógeno abajo), y entre ellos la barrera se atraviesa por efecto túnel cuántico. La energía del estado simétrico \(|II\rangle\) es \(E_0 - A\) (la más baja), y la del estado antisimétrico \(|I\rangle\) es \(E_0 + A\) (la más alta).
✅ Verificación de comprensión: La frecuencia de microondas correspondiente al desdoblamiento túnel \(2A\) de la molécula de amoníaco es aproximadamente 24,000 MHz. Si no hubiera efecto túnel (\(A = 0\)), ¿cómo serían los niveles de energía?
Respuesta
Si \(A = 0\) entonces \(E_I = E_{II} = E_0\), y los dos niveles de energía se superponen completamente (están degenerados). Si el átomo de nitrógeno no puede atravesar el plano, las configuraciones "arriba" y "abajo" son independientes e indistinguibles, y no surge ninguna diferencia de energía.
📝 Ejercicios:
- Verificar la derivación del autovector correspondiente al autovalor \(E_I = E_0 + A\), partiendo de la ecuación (6.9b). → Problema B-4. Verificación de la ortogonalidad de los vectores propios
6.5 Evolución temporal de los estados estacionarios¶
🟡 Lina: Ya que conocemos los autovalores y autovectores, podemos escribir la evolución temporal de los estados estacionarios. Un sistema que en el instante inicial \(t = 0\) está en el autoestado \(|I\rangle\), en el instante \(t\) está en:
Igualmente, para el autoestado \(|II\rangle\):
🔵 Kai: \(e^{-iEt/\hbar}\) siempre tiene valor absoluto 1, ¿verdad? Entonces \(|C_1|^2\) y \(|C_2|^2\) no cambian con el tiempo.
🟡 Lina: Exacto. En los estados estacionarios, las probabilidades de encontrar el sistema en \(|1\rangle\) o \(|2\rangle\) no dependen del tiempo. Por eso se llaman "estacionarios".
✅ Verificación de comprensión: En los estados estacionarios las amplitudes tienen una dependencia temporal \(e^{-iEt/\hbar}\), ¿por qué se llaman "estacionarios"?
Respuesta
\(e^{-iEt/\hbar}\) es un factor de fase con valor absoluto siempre igual a 1, por lo que las probabilidades \(|C_1|^2\) y \(|C_2|^2\) no dependen del tiempo. Como las magnitudes físicas observables (probabilidades) no cambian, se denominan "estacionarios". Lo que cambia es solo la fase de las amplitudes.
⚪ Mei: Por ejemplo, estando en el estado \(|II\rangle\), la probabilidad de encontrarlo en \(|1\rangle\) es \(|1/\sqrt{2}|^2 = 1/2\). La probabilidad de encontrarlo en \(|2\rangle\) también es \(1/2\). La probabilidad de que el nitrógeno esté arriba y la de que esté abajo son siempre iguales a la mitad, y esto no cambia con el tiempo.
🟡 Lina: Sin embargo, la fase de las amplitudes sigue rotando. \(e^{-iE_I t/\hbar}\) y \(e^{-iE_{II} t/\hbar}\) rotan con frecuencias angulares distintas, así que si creamos una superposición que no sea un estado estacionario — ocurre algo interesante.
6.6 Oscilaciones cuánticas — Evolución temporal de las amplitudes según la condición inicial¶
🟡 Lina: Llegamos al punto culminante de este capítulo. Consideremos la condición inicial de que en \(t = 0\) el nitrógeno está "arriba" — es decir, \(|\psi(0)\rangle = |1\rangle\).
🔵 Kai: \(|1\rangle\) no es un autoestado, ¿verdad? Necesitamos escribirlo como superposición de \(|I\rangle\) y \(|II\rangle\).
🟡 Lina: Así es. Sumemos las ecuaciones (6.15a) y (6.15b) para obtener \(|1\rangle\):
Por lo tanto:
Igualmente, calculando \(|II\rangle - |I\rangle\):
Por lo tanto:
🟡 Lina: Esto es un cambio de base. Tanto \(\{|1\rangle, |2\rangle\}\) como \(\{|I\rangle, |II\rangle\}\) son bases del sistema de 2 estados, pero la segunda consiste en autoestados del hamiltoniano, así que la llamamos base de energía.
⚪ Mei: Es simplemente cambiar el punto de vista de un mismo estado — verlo desde la base de posición o desde la base de energía.
🟡 Lina: Así es. Ahora, reescribimos \(|\psi(0)\rangle = |1\rangle\) usando la ecuación (6.17a) y añadimos a cada autoestado el factor de fase de evolución temporal. ¿Por qué podemos hacer esto? Porque la ecuación (6.2) es lineal. En ecuaciones lineales, "la superposición de soluciones es también solución" — así que si encontramos la evolución temporal de \(|I\rangle\) y la de \(|II\rangle\) por separado y las sumamos, obtenemos la evolución temporal total:
Devolviendo \(|I\rangle\) y \(|II\rangle\) a la base original:
Reuniendo el coeficiente de \(|1\rangle\): la contribución de \(|I\rangle\) es \(\frac{1}{\sqrt{2}} \cdot \frac{1}{\sqrt{2}} = \frac{1}{2}\), y la de \(|II\rangle\) también es \(\frac{1}{\sqrt{2}} \cdot \frac{1}{\sqrt{2}} = \frac{1}{2}\):
Igualmente, hallemos el coeficiente de \(|2\rangle\). Con el mismo método que para \(C_1\), recogemos de cada autoestado el coeficiente de \(|2\rangle\). Como \(|I\rangle = \frac{1}{\sqrt{2}}(|1\rangle - |2\rangle)\), el coeficiente de \(|2\rangle\) en \(|I\rangle\) es \(-1/\sqrt{2}\). En la ecuación (6.18), \(|I\rangle\) tiene un factor \(1/\sqrt{2}\) delante, así que la contribución de \(|I\rangle\) a \(|2\rangle\) es \(\frac{1}{\sqrt{2}} \times (-\frac{1}{\sqrt{2}}) = -\frac{1}{2}\). Igualmente, como \(|II\rangle = \frac{1}{\sqrt{2}}(|1\rangle + |2\rangle)\), el coeficiente de \(|2\rangle\) en \(|II\rangle\) es \(+1/\sqrt{2}\), y en (6.18) \(|II\rangle\) también tiene un factor \(1/\sqrt{2}\) delante, así que la contribución de \(|II\rangle\) es \(\frac{1}{\sqrt{2}} \times \frac{1}{\sqrt{2}} = +\frac{1}{2}\). Por lo tanto (escribiendo en orden la contribución de \(|I\rangle\) y la de \(|II\rangle\)):
(La última igualdad solo intercambia el orden de los dos términos con \(-a + b = b - a\). La razón de reescribirlo así es que al sustituir \(E_I = E_0 + A\), \(E_{II} = E_0 - A\), se tiene \(e^{-iE_{II} t/\hbar} - e^{-iE_I t/\hbar} = e^{-i(E_0-A)t/\hbar} - e^{-i(E_0+A)t/\hbar}\), y al extraer el factor común \(e^{-iE_0 t/\hbar}\) aparece la forma \(e^{+iAt/\hbar} - e^{-iAt/\hbar}\) — que por la fórmula de Euler se convierte en \(\sin\).)
🔵 Kai: Suma y diferencia de dos exponenciales complejas... ¿Es algo como "batidos"?
🟡 Lina: ¡Exactamente! Extraigamos el factor común. Sustituyendo \(E_I = E_0 + A\), \(E_{II} = E_0 - A\) en (6.19a):
Dentro del paréntesis tenemos la forma \(e^{-iAt/\hbar} + e^{+iAt/\hbar}\). Igualmente, para \(C_2\):
⚪ Mei: \(C_1\) tiene la forma de "suma" y \(C_2\) la forma de "diferencia" — parece que van a salir funciones trigonométricas.
🟡 Lina: Aquí usamos la fórmula de Euler. De \(e^{i\theta} = \cos\theta + i\sin\theta\) introducida en Cap. 4, se derivan dos fórmulas útiles:
Para \(C_1\), dentro del paréntesis tenemos \(e^{-iAt/\hbar} + e^{+iAt/\hbar} = 2\cos(At/\hbar)\), y multiplicando por \(1/2\) queda \(\cos(At/\hbar)\). Para \(C_2\), dentro del paréntesis tenemos \(-e^{-iAt/\hbar} + e^{+iAt/\hbar}\). Intercambiando el orden de los términos es \(e^{+iAt/\hbar} - e^{-iAt/\hbar}\) — que es exactamente la segunda fórmula con \(\theta = At/\hbar\). Entonces \(e^{+iAt/\hbar} - e^{-iAt/\hbar} = 2i\sin(At/\hbar)\), y multiplicando por \(1/2\) queda \(i\sin(At/\hbar)\).
🔵 Kai: ¡Ooh, se separa limpiamente en \(\cos\) e \(i\sin\)!
🟡 Lina: Exacto. Y como \(\cos\) y \(\sin\) están desfasadas en \(\pi/2\), las dos amplitudes alternan entre hacerse grandes y pequeñas — esta es la esencia de las oscilaciones cuánticas. Resumiendo:
🟡 Lina: Calculemos las probabilidades:
🔵 Kai: ¡¡¡Ohhh!!! La probabilidad va y viene con \(\cos^2\) y \(\sin^2\)... ¿Eso significa que el átomo de nitrógeno oscila periódicamente entre arriba y abajo?
🟡 Lina: Exacto. El átomo de nitrógeno, que clásicamente no podría superar la barrera, en mecánica cuántica va y viene probabilísticamente — esa es la esencia de las oscilaciones cuánticas. Verifiquémoslo concretamente. En \(t = 0\), \(\cos^2(0) = 1\) así que \(P_1 = 1\) y el nitrógeno ciertamente está arriba. Con el paso del tiempo, \(P_1\) disminuye.
🔵 Kai: \(\cos^2\) se anula cuando... \(At/\hbar = \pi/2\), así que en \(t = \pi\hbar/(2A)\) tenemos \(P_1 = 0\). ¿¡Eso significa que hay un instante en que el nitrógeno está completamente "abajo"!? ¡Se supone que no puede superar la barrera!
⚪ Mei: Sí. En ese momento \(P_2 = \sin^2(\pi/2) = 1\), así que está abajo con probabilidad del 100%. Y luego vuelve — es una oscilación periódica.
🟡 Lina: Esto se llama oscilación cuántica (quantum oscillation), u oscilación de Rabi. Hallemos la frecuencia angular. Reescribiendo \(P_1(t) = \cos^2(At/\hbar)\) con la fórmula del ángulo mitad \(\cos^2\theta = \frac{1}{2}(1 + \cos 2\theta)\), obtenemos \(P_1(t) = \frac{1}{2}(1 + \cos(2At/\hbar))\). Viendo esta expresión, la probabilidad oscila alrededor del valor constante \(1/2\) con \(\cos(2At/\hbar)\). Como el argumento del \(\cos\) es \(2At/\hbar\), la frecuencia angular de oscilación de la probabilidad es:
El período de oscilación es:
Durante un período \(T\), el átomo de nitrógeno completa un ciclo "arriba→abajo→arriba". El gráfico de esta oscilación se muestra en Fig. 6.4「Evolución temporal de la probabilidad por oscilaciones de Rabi」.
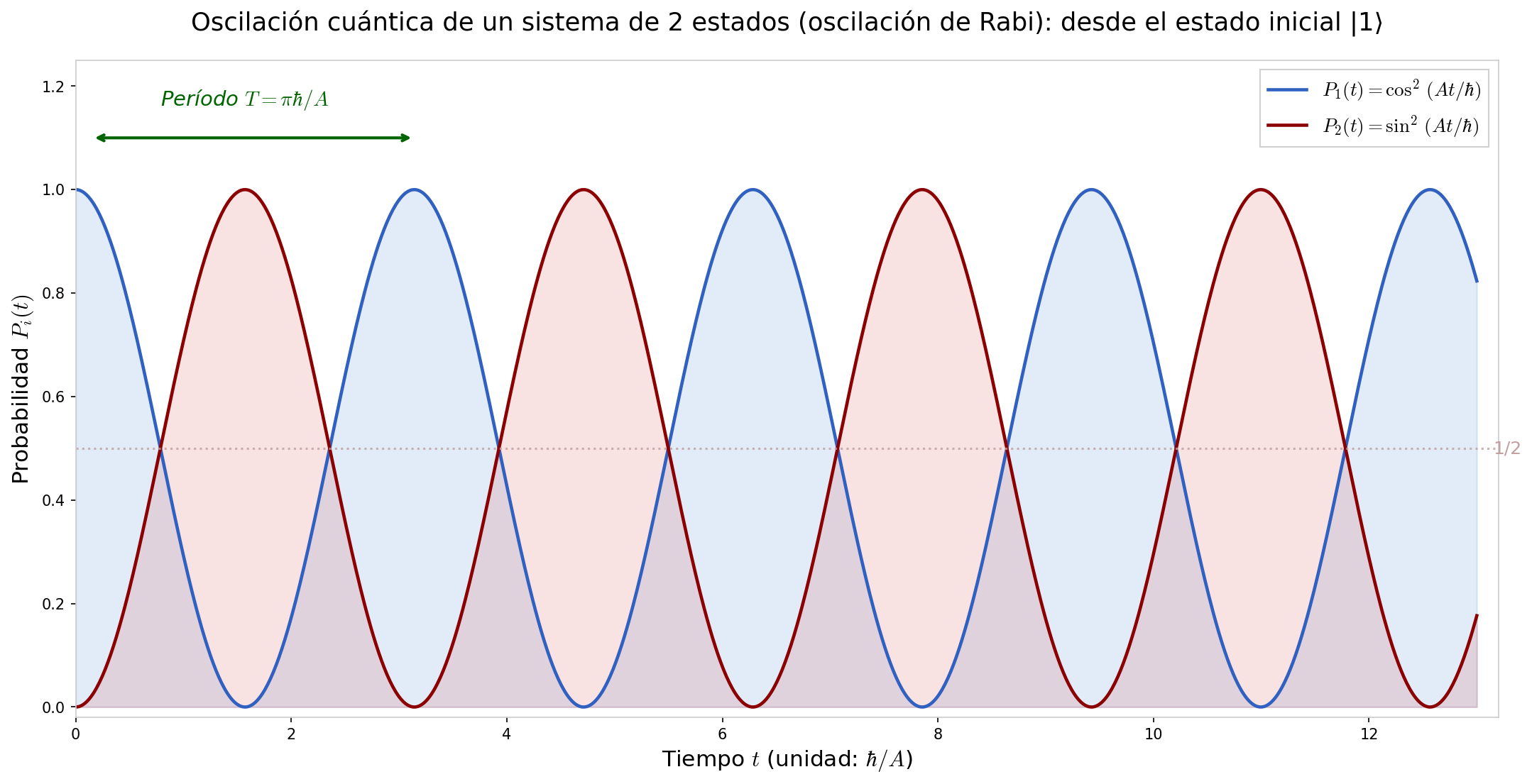
Fig. 6.4: Evolución temporal de la probabilidad por oscilaciones de Rabi. Oscilación de Rabi desde el estado inicial \(|\psi(0)\rangle = |1\rangle\). \(P_1(t) = \cos^2(At/\hbar)\) (azul) y \(P_2(t) = \sin^2(At/\hbar)\) (rojo). La probabilidad oscila completamente entre los dos estados, y \(P_1 + P_2 = 1\) se cumple siempre.
⚪ Mei: Como la probabilidad oscila con \(\cos^2\) y \(\sin^2\), \(P_1 + P_2 = \cos^2 + \sin^2 = 1\) se cumple siempre. Se verifica la conservación de la probabilidad.
🔵 Kai: Un momento. Se supone que clásicamente el átomo de nitrógeno no tiene suficiente energía para "filtrarse" a través del plano de hidrógenos, ¿verdad? Y sin embargo la probabilidad crece con \(\sin^2\)... Pero si la barrera se hiciera más alta, ¿\(A\) se haría más pequeña? Es decir, ¿en el límite de barrera infinitamente alta desaparece el efecto túnel y la oscilación se detiene?
🟡 Lina: Buena pregunta. Exacto. Cuanto más alta y gruesa es la barrera, \(A\) decrece exponencialmente. Clásicamente, el átomo de nitrógeno está bloqueado por la barrera de potencial y no puede pasar al otro lado. Pero en mecánica cuántica, mientras la amplitud \(A\) de "tunelaje" no sea cero, la oscilación ocurre. Si \(A\) es pequeña la oscilación es lenta, pero con una barrera finita nunca es cero.
🔵 Kai: "Decrece exponencialmente" significa que con solo engrosar un poco la barrera, \(A\) se reduce drásticamente... Entonces, a escalas cotidianas donde la barrera es enormemente gruesa, ¿la probabilidad de atravesar la pared es prácticamente cero?
🟡 Lina: Exacto. Para objetos macroscópicos, la barrera es enormemente gruesa comparada con la escala atómica, así que \(A\) se vuelve astronómicamente pequeña. Pero a la inversa, mientras \(A\) sea finita — es decir, mientras la barrera tenga altura y anchura finitas — la oscilación cuántica nunca es cero. "Detenerse completamente" solo ocurre en el límite de barrera infinita. Esta es la diferencia esencial con la mecánica clásica.
⚪ Mei: Es decir, lo que en mecánica clásica es "una pared infranqueable", en mecánica cuántica se convierte en "una pared difícil de franquear" — es cuestión de grado, y en principio nunca es cero.
✅ Verificación de comprensión: ¿Cómo se expresa la frecuencia angular \(\omega_0\) de la oscilación de Rabi? Además, ¿la oscilación se vuelve más rápida o más lenta cuando la amplitud de túnel \(A\) es mayor?
Respuesta
\(\omega_0 = 2A/\hbar\). Cuanto mayor es \(A\), mayor es la frecuencia angular, así que la oscilación se vuelve más rápida. Cuanto más fuerte es el efecto túnel (más fácil es atravesar la barrera), más frecuente es el ir y venir del átomo de nitrógeno entre arriba y abajo.
✅ Verificación de comprensión: Si el estado inicial fuera \(|\psi(0)\rangle = |2\rangle\) (nitrógeno "abajo"), ¿cómo serían \(P_1(t)\) y \(P_2(t)\)?
Respuesta
De la ecuación (6.17b), \(|2\rangle = \frac{1}{\sqrt{2}}(-|I\rangle + |II\rangle)\). Haciendo un cálculo similar se obtiene \(P_1(t) = \sin^2(At/\hbar)\), \(P_2(t) = \cos^2(At/\hbar)\). Es decir, solo se intercambia la condición inicial, y el patrón de oscilación es el mismo (\(\cos\) y \(\sin\) se intercambian).
📝 Ejercicios:
- Cuando el estado inicial es \(|\psi(0)\rangle = |II\rangle\) (autoestado de baja energía), calcular \(P_1(t)\) y \(P_2(t)\) y verificar que no dependen del tiempo. → Problema B-5. Cálculo de la dependencia temporal de la probabilidad
6.7 Por qué el hamiltoniano está en el centro — Repaso de lo aprendido¶
🔵 Kai: Llegados a este punto, la matriz hamiltoniana es realmente poderosa. Con solo escribir una matriz, se obtienen los niveles de energía, los estados estacionarios y la evolución temporal.
🟡 Lina: Así es. "Resolver un problema" en mecánica cuántica se reduce, en esencia, a escribir el hamiltoniano y encontrar sus autovalores y autovectores. El procedimiento resumido es:
- Elegir la base: Establecer una base (basis) \(\{|1\rangle, |2\rangle, \ldots\}\) físicamente natural
- Determinar la matriz hamiltoniana: Determinar \(H_{ij}\) usando simetrías y consideraciones físicas
- Resolver el problema de autovalores: \(H|E_n\rangle = E_n|E_n\rangle\) — obtener los niveles de energía \(E_n\) y los autoestados \(|E_n\rangle\)
- Expandir la condición inicial en autoestados: \(|\psi(0)\rangle = \sum_n c_n |E_n\rangle\)
- Escribir la evolución temporal: \(|\psi(t)\rangle = \sum_n c_n\,e^{-iE_n t/\hbar}|E_n\rangle\)
Tabla 6.2: Los 5 pasos para resolver un problema de mecánica cuántica
| Paso | Operación | Ejemplo concreto con la molécula de amoníaco |
|---|---|---|
| 1. Elegir la base | Enumerar estados físicamente naturales | \(\|1\rangle\) (N arriba), \(\|2\rangle\) (N abajo) |
| 2. Determinar el hamiltoniano | Usar simetrías y consideraciones físicas | \(H = \begin{pmatrix} E_0 & -A \\\\ -A & E_0 \end{pmatrix}\) |
| 3. Resolver el problema de autovalores | \(\det(H - E\mathbb{1}) = 0\) | \(E_I = E_0 + A\), \(E_{II} = E_0 - A\) |
| 4. Expandir la condición inicial | \(c_n = \langle E_n\|\psi(0)\rangle\) | \(\|1\rangle = \frac{1}{\sqrt{2}}(\|I\rangle + \|II\rangle)\) |
| 5. Escribir la evolución temporal | Añadir \(e^{-iE_n t/\hbar}\) a cada autoestado | \(P_1(t) = \cos^2(At/\hbar)\) (oscilación de Rabi) |
⚪ Mei: Esta receta no se limita a sistemas de 2 estados, tiene la misma estructura para sistemas con más estados.
🟡 Lina: Exacto. A partir de Cap. 7 introduciremos la función de onda y la ecuación de Schrödinger, pero la estructura lógica fundamental es la misma que vimos aquí. Los autovalores del hamiltoniano son las energías, los autoestados son los estados estacionarios, y un estado general evoluciona como superposición de autoestados — este esqueleto recorre toda la mecánica cuántica.
🔵 Kai: Practicar con el sistema de 2 estados parece que facilita lo que viene después... Pero si hubiera 3 o más estados, ¿no se volvería muy difícil encontrar los autovalores? Saldrían ecuaciones de tercer grado y así.
🟡 Lina: Buena preocupación. Efectivamente, para un sistema de \(N\) estados hay que resolver una ecuación de grado \(N\). Pero en muchos casos, se pueden usar simetrías para descomponer el problema en bloques más pequeños. Eso lo veremos en capítulos posteriores.
🔵 Kai: La simetría vuelve a aparecer. En el amoníaco también fue la simetría la que determinó \(H_{11} = H_{22}\)... dicho al revés, ¿para sistemas sin simetría sería mucho más difícil determinar el hamiltoniano?
🟡 Lina: Exacto. Cuanta menos simetría tiene un sistema, más datos experimentales u otras consideraciones teóricas se necesitan para determinar los componentes del hamiltoniano. Pero inversamente, encontrar simetrías es el arma más poderosa para resolver problemas — esta es una lección que aplica no solo a la mecánica cuántica sino a toda la física.
🔵 Kai: "Encontrar simetrías es un arma"... Ciertamente, en el amoníaco, una sola simetría arriba-abajo determinó la mitad de la matriz.
✅ Verificación de comprensión: Describe en 5 pasos el procedimiento básico para "resolver un problema" en mecánica cuántica.
Respuesta
(1) Elegir la base (basis), (2) determinar la matriz hamiltoniana, (3) resolver el problema de autovalores para obtener los niveles de energía y autoestados, (4) expandir la condición inicial en autoestados, (5) escribir la evolución temporal añadiendo el factor de fase \(e^{-iE_n t/\hbar}\) a cada autoestado.
6.8 La molécula de amoníaco en un campo eléctrico — Cuando la simetría se rompe¶
🟡 Lina: Avancemos un paso más y consideremos el caso en que se aplica un campo eléctrico externo a la molécula de amoníaco.
🔵 Kai: ¿Qué cambia al aplicar un campo eléctrico?
🟡 Lina: La molécula de amoníaco posee un momento dipolar eléctrico (electric dipole moment) \(\mu\). El momento dipolar eléctrico es una cantidad que representa la magnitud y dirección del desplazamiento entre los centros de carga positiva y negativa dentro de la molécula. Cuantitativamente, cuando una carga positiva \(q\) y una carga negativa \(-q\) están separadas una distancia \(d\), la magnitud del momento dipolar se define como \(\mu = qd\). Sus dimensiones son \([\text{carga}] \times [\text{longitud}]\), y la unidad SI es C·m (coulomb·metro).
🔵 Kai: Lo positivo y lo negativo están desplazados... ¿Concretamente cómo es?
🟡 Lina: El átomo de nitrógeno atrae a los electrones más fuertemente que los átomos de hidrógeno. Por eso el lado del nitrógeno queda ligeramente negativo (\(-\)) y el lado de los hidrógenos queda ligeramente positivo (\(+\)). La dirección del momento dipolar se define como la dirección desde la carga negativa hacia la carga positiva. Recuérdalo como "una flecha que apunta desde \(-\) hacia \(+\)". Mira de nuevo Fig. 6.1「Estructura de la molécula de amoníaco」 — en la configuración donde el nitrógeno (\(-\)) está arriba y los hidrógenos (\(+\)) están abajo, la flecha va de arriba (\(-\)) hacia abajo (\(+\)), así que el dipolo apunta "hacia abajo". En la configuración con el nitrógeno "abajo", es al revés y el dipolo apunta "hacia arriba".
Y ahora hablemos de la energía cuando un dipolo se coloca en un campo eléctrico. La carga positiva es atraída en la dirección del campo, y la carga negativa es atraída en dirección opuesta. Cuando la flecha del dipolo apunta en la misma dirección que el campo — es decir, cuando el lado de la carga positiva está en la dirección del campo — es lo más estable y la energía es mínima. Cuando apunta en dirección opuesta es lo más inestable y la energía es máxima. Esto se expresa como "el dipolo está alineado con el campo".
🔵 Kai: A ver, déjame ordenar. Cuando el nitrógeno está "arriba", la carga negativa (lado del nitrógeno) está arriba y la positiva (lado de los hidrógenos) está abajo... la flecha va del \(-\) de arriba al \(+\) de abajo. Es decir, el dipolo apunta "hacia abajo". Si el nitrógeno está "abajo", la carga negativa está abajo y la positiva arriba, así que la flecha va de abajo hacia arriba y es "hacia arriba".
⚪ Mei: "La flecha del dipolo apunta de \(-\) a \(+\)" — con solo recordar eso, una vez decidida la posición del nitrógeno, queda determinada la dirección de la flecha.
🟡 Lina: Exacto. Lo habéis repasado con calma. El punto clave es que "la dirección de la flecha" y "la posición del nitrógeno" son siempre opuestas — nitrógeno arriba significa flecha abajo, nitrógeno abajo significa flecha arriba. Grábate esta correspondencia.
⚪ Mei: Cuando la posición del nitrógeno se invierte, la dirección del momento dipolar también se invierte.
🟡 Lina: Así es. Supongamos que aplicamos un campo eléctrico externo \(\mathcal{E}\) "hacia arriba". Cuando un dipolo se coloca en un campo eléctrico, el lado de la carga positiva es atraído en la dirección del campo y el de la negativa en dirección opuesta. Cuando el dipolo está alineado con el campo es "estable" y la energía es baja; cuando está en dirección opuesta es "inestable" y la energía es alta. Cuantitativamente, la energía de interacción entre el momento dipolar y el campo eléctrico es \(U = -\mu\mathcal{E}\cos\theta\) (donde \(\theta\) es el ángulo entre el dipolo y el campo). Para explicar brevemente el origen de esta fórmula: la carga positiva \(+q\) experimenta una fuerza \(F = qE\) en la dirección del campo, y la carga negativa \(-q\) experimenta la misma magnitud de fuerza en dirección opuesta. Cuando el dipolo forma un ángulo \(\theta\) con el campo, la "longitud efectiva" en la dirección del campo es \(d\cos\theta\), así que el trabajo (energía) es \(-qd\mathcal{E}\cos\theta = -\mu\mathcal{E}\cos\theta\). Cuando están alineados (\(\theta = 0°\)) la energía es mínima \(U = -\mu\mathcal{E}\); cuando están en dirección opuesta (\(\theta = 180°\)) la energía es máxima \(U = +\mu\mathcal{E}\).
🔵 Kai: Cuando se alinea la energía baja, y cuando es opuesto la energía sube... se parece a un imán que tiende a alinearse con el campo magnético.
🟡 Lina: Buena analogía. Verifiquémoslo concretamente. En el estado \(|1\rangle\) (nitrógeno arriba), como confirmamos antes, el dipolo apunta hacia abajo. El campo es hacia arriba, así que dipolo y campo son opuestos (\(\theta = 180°\)) y \(U = +\mu\mathcal{E}\) (energía alta). En el estado \(|2\rangle\) (nitrógeno abajo), el dipolo apunta hacia arriba, en la misma dirección que el campo (\(\theta = 0°\)) y \(U = -\mu\mathcal{E}\) (energía baja).
🔵 Kai: ¡Entonces \(H_{11} \neq H_{22}\)! ¡La simetría se rompe!
🟡 Lina: Exacto. El hamiltoniano en presencia de campo eléctrico es:
Aquí asumimos que la amplitud de túnel \(-A\) apenas se ve afectada por el campo eléctrico.
🔵 Kai: Me gustaría ver en un gráfico cómo se mueven los niveles de energía cuando se varía el campo \(\mathcal{E}\).
🟡 Lina: Buena propuesta. Mira Fig. 6.5「Niveles de energía en un campo eléctrico」. Si \(A = 0\) (sin efecto túnel), los dos niveles de energía varían linealmente y se cruzan en \(\mu\mathcal{E} = 0\) (líneas discontinuas). Pero si \(A \neq 0\), en el punto donde se cruzarían, los niveles se "repelen" — esto se llama cruce evitado (avoided crossing). Calcularemos los autovalores enseguida, pero adelantando el resultado, la separación entre niveles es \(2\sqrt{A^2 + \mu^2\mathcal{E}^2}\). Mientras \(A \neq 0\) esto nunca es cero — es decir, las dos líneas nunca se cruzan. El punto de máximo acercamiento (en \(\mathcal{E} = 0\)) tiene una separación de \(2A\).
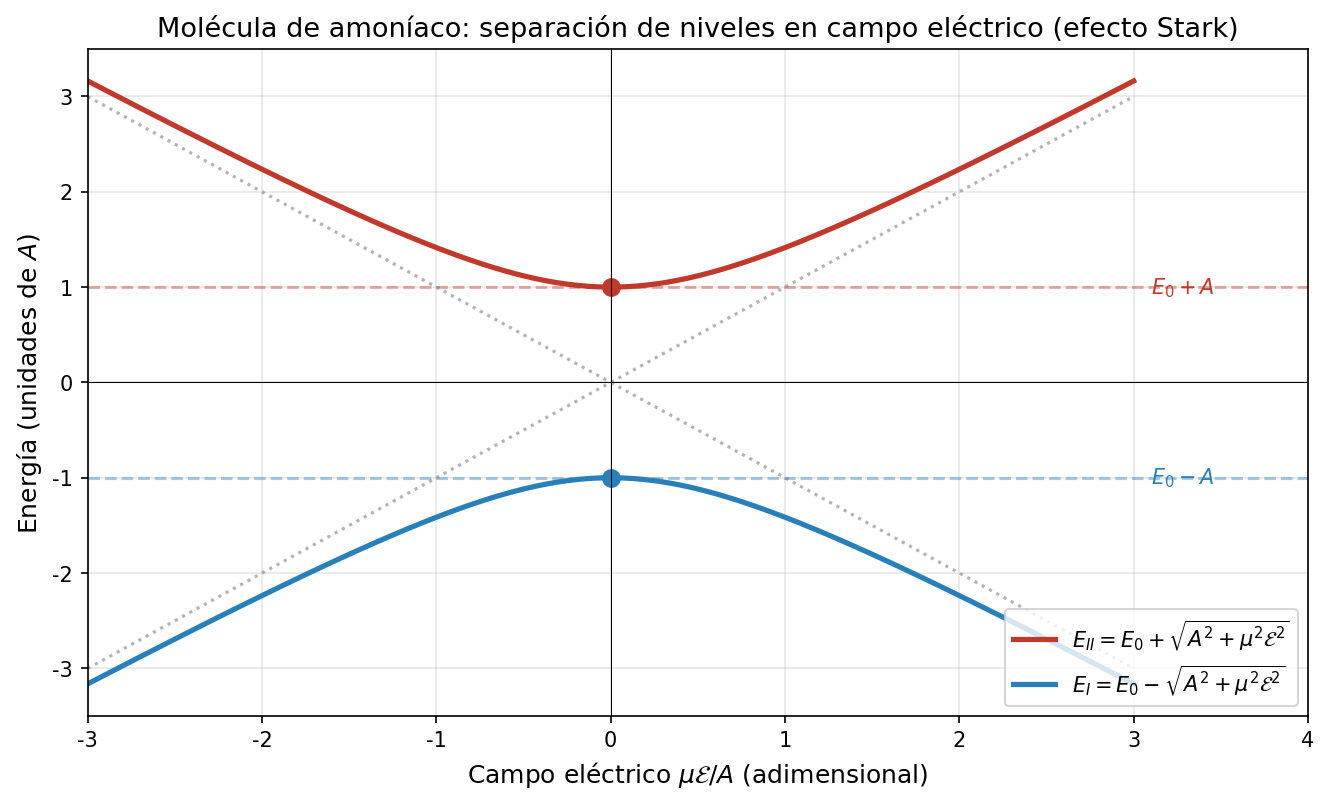
Fig. 6.5: Niveles de energía en un campo eléctrico. El eje horizontal es \(\mu\mathcal{E}/A\), el eje vertical es la energía. Las líneas discontinuas son el caso \(A = 0\) (sin efecto túnel), donde dos rectas se cruzan. Las líneas continuas son el caso \(A \neq 0\), donde el cruce se evita (cruce evitado) y la separación mínima es \(2A\).
Autovalores en un campo eléctrico¶
🟡 Lina: Calculemos los autovalores. Para un hamiltoniano general de 2 estados
los autovalores son:
🔵 Kai: ¿De dónde sale esto?
🟡 Lina: Del mismo procedimiento que en la sección anterior. Escribiendo \(\det(H - E\mathbb{1}) = 0\) con componentes generales: \((H_{11} - E)(H_{22} - E) - H_{12}H_{21} = 0\) — simplemente usando la definición del determinante \(ad - bc\) con \(a = H_{11} - E\), \(b = H_{12}\), \(c = H_{21}\), \(d = H_{22} - E\). Expandimos. Usando la hermiticidad \(H_{12}H_{21} = H_{12}H_{12}^* = |H_{12}|^2\):
Esto es una ecuación de segundo grado en \(E\), así que usamos la fórmula que aprendiste en la preparatoria: \(E = \frac{(H_{11}+H_{22}) \pm \sqrt{(H_{11}+H_{22})^2 - 4(H_{11}H_{22} - |H_{12}|^2)}}{2}\). Simplificando el discriminante: \((H_{11}+H_{22})^2 - 4H_{11}H_{22} + 4|H_{12}|^2 = (H_{11}-H_{22})^2 + 4|H_{12}|^2\), de donde se obtiene la ecuación (6.25). Esta es una fórmula general aplicable a cualquier hamiltoniano de 2 estados, no solo a la molécula de amoníaco.
⚪ Mei: Es decir, conociendo solo \(H_{11}\), \(H_{22}\) y \(H_{12}\), se obtienen inmediatamente los autovalores.
🟡 Lina: Para la molécula de amoníaco, sustituyendo \(H_{11} + H_{22} = 2E_0\), \(H_{11} - H_{22} = 2\mu\mathcal{E}\), \(|H_{12}|^2 = A^2\):
🔵 Kai: Cuando \(\mathcal{E} = 0\), \(\sqrt{A^2} = A\), así que recuperamos \(E_I = E_0 + A\), \(E_{II} = E_0 - A\).
🟡 Lina: Así es. A medida que el campo se hace más fuerte, la separación entre niveles \(E_I - E_{II} = 2\sqrt{A^2 + \mu^2\mathcal{E}^2}\) se amplía.
Comportamiento en los límites¶
🟡 Lina: Examinemos dos límites.
Campo débil (\(\mu\mathcal{E} \ll A\)):
Aquí usamos la aproximación \(\sqrt{1+x} \approx 1 + x/2\) (cuando \(x \ll 1\)). Esta es la expansión de Taylor a primer orden de \((1+x)^{1/2}\) — es decir, "cuando \(x\) es suficientemente pequeño, ignoramos los términos de orden \(x^2\) y superiores y dejamos solo el término de primer orden". Probando con \(x = 0.01\): \(\sqrt{1.01} = 1.00499\ldots \approx 1 + 0.005 = 1.005\), que es una buena aproximación, ¿verdad?
Por lo tanto:
⚪ Mei: El cambio de energía es proporcional a \(\mathcal{E}^2\). Que sea al cuadrado y no a la primera potencia es interesante.
🟡 Lina: Así es. Cuando el cambio de energía es proporcional a \(\mathcal{E}^2\), el coeficiente de proporcionalidad se llama polarizabilidad (polarizability). Aquí, \(\mu^2/(2A)\) desempeña ese papel. Cuanto menor es \(A\), mayor es la polarizabilidad — es decir, más sensible al campo. Intuitivamente, que \(A\) sea pequeño significa que los dos niveles están próximos. Cuanto más cercanos están los niveles, más fácilmente se mezclan los estados ante una perturbación externa (campo eléctrico) y más se desplaza la energía. La molécula de amoníaco tiene \(A\) muy pequeño (escala de energía de microondas), por lo que su polarizabilidad es anormalmente grande.
🔵 Kai: Niveles cercanos significan sensibilidad al campo... es como si un equilibrio inestable se moviera mucho con solo un empujón.
🟡 Lina: Campo fuerte (\(\mu\mathcal{E} \gg A\)):
🔵 Kai: ¿Esto significa que \(A\) deja de importar?
🟡 Lina: Así es. Cuando el campo es muy fuerte, la diferencia de energía "arriba" vs "abajo" \(2\mu\mathcal{E}\) domina sobre la amplitud de túnel \(A\). El átomo de nitrógeno queda "fijado" en el lado de menor energía, y el efecto de tunelaje al otro lado se vuelve casi invisible.
🔵 Kai: Un campo fuerte "mata" el efecto túnel. Intuitivamente tiene sentido. Es como si la pendiente de energía fuera tan pronunciada que filtrase no tiene sentido.
🟡 Lina: Resumamos los dos límites en una tabla.
Tabla 6.3: Comparación del comportamiento según la intensidad del campo
| Campo débil (\(\mu\mathcal{E} \ll A\)) | Campo fuerte (\(\mu\mathcal{E} \gg A\)) | |
|---|---|---|
| Dependencia de la energía con el campo | Proporcional a \(\mathcal{E}^2\) (segundo orden) | Proporcional a \(\mathcal{E}\) (primer orden) |
| Separación entre niveles | \(\approx 2A\) (casi constante) | \(\approx 2\mu\mathcal{E}\) (proporcional al campo) |
| Carácter de los autoestados | Simétrico/antisimétrico (\(\|1\rangle \pm \|2\rangle\)) | Casi \(\|1\rangle\) y \(\|2\rangle\) (localizados) |
| Influencia del efecto túnel | Dominante (ocurren oscilaciones cuánticas) | Suprimido (nitrógeno fijado en un lado) |
| Descripción física | El nitrógeno va y viene arriba-abajo | El nitrógeno permanece en el lado de menor energía |
✅ Verificación de comprensión: En el límite de campo débil (\(\mu\mathcal{E} \ll A\)), ¿a qué potencia de \(\mathcal{E}\) es proporcional la dependencia de los niveles de energía con el campo? ¿Cómo se llama el coeficiente de proporcionalidad?
Respuesta
En campo débil, el cambio de energía es proporcional a \(\mathcal{E}^2\) (efecto de segundo orden). El coeficiente de proporcionalidad \(\mu^2/(2A)\) se llama polarizabilidad (polarizability). Cuanto menor es \(A\), mayor es la polarizabilidad y más sensible es al campo eléctrico.
✅ Verificación de comprensión: Cuando el campo \(\mathcal{E}\) se aumenta gradualmente, ¿cómo cambia la separación entre los niveles de energía \(E_I\) y \(E_{II}\)?
Respuesta
\(E_I - E_{II} = 2\sqrt{A^2 + \mu^2\mathcal{E}^2}\). En \(\mathcal{E} = 0\) vale \(2A\); a medida que \(\mathcal{E}\) aumenta, crece monótonamente; y para \(\mathcal{E} \gg A/\mu\) se aproxima a \(2\mu\mathcal{E}\). La separación entre niveles se amplía con el campo y nunca se reduce.
📝 Ejercicios:
- Derivar la ecuación (6.25) a partir de la fórmula de resolución de ecuaciones de segundo grado. → Problema M-1. Derivación de la hermiticidad a partir de la conservación de la probabilidad
6.9 Principio del máser de amoníaco — Transiciones entre estados por un campo eléctrico oscilante¶
🟡 Lina: Finalmente, toquemos el principio de funcionamiento del máser de amoníaco (ammonia maser). MASER es el acrónimo de Microwave Amplification by Stimulated Emission of Radiation (amplificación de microondas por emisión estimulada de radiación).
🔵 Kai: En Cap. 1 se habló de la emisión estimulada de Einstein (1917), ¿verdad? ¿Está relacionado?
🟡 Lina: Exactamente. El máser es la realización de la emisión estimulada predicha por Einstein, usando el sistema de 2 niveles de la molécula de amoníaco. Fue el primer dispositivo puesto en funcionamiento por Townes y colaboradores en 1954, y se convirtió en el precursor del láser (LASER).
Separación de moléculas¶
🟡 Lina: Para el funcionamiento del máser se necesita una preparación. Primero se crea un haz de moléculas de amoníaco y se separan las moléculas en estado \(|I\rangle\) (alta energía) de las de estado \(|II\rangle\) (baja energía).
🔵 Kai: ¿Cómo se separan?
🟡 Lina: Se usa un campo eléctrico no uniforme. Mira la ecuación (6.27). En la aproximación de campo débil:
- La energía del estado \(|I\rangle\) aumenta con \(\mathcal{E}^2\)
- La energía del estado \(|II\rangle\) disminuye con \(\mathcal{E}^2\)
En la física del bachillerato aprendiste que "un objeto experimenta una fuerza en la dirección en que la energía potencial disminuye", ¿verdad? Es decir, la fuerza es \(F = -dU/dx\) — en la dirección donde la energía aumenta con la posición, se ejerce una fuerza que la empuja de vuelta. En un campo no uniforme, la intensidad del campo \(\mathcal{E}\) varía según la posición, así que la energía de la molécula también varía con la posición. Las moléculas en estado \(|I\rangle\) tienen energía que aumenta con \(\mathcal{E}^2\), así que donde el campo es más intenso la energía es mayor — por lo tanto experimentan una fuerza hacia la región de campo débil. Inversamente, las moléculas en estado \(|II\rangle\) tienen energía que disminuye con \(\mathcal{E}^2\), así que donde el campo es más intenso la energía es menor — por lo tanto experimentan una fuerza hacia la región de campo fuerte. Así el haz se divide en dos.
🔵 Kai: ¡Ah, esto se parece al experimento de Stern-Gerlach de Cap. 5! Allí se separaban espines con un campo magnético no uniforme.
🟡 Lina: Buena conexión. Es exactamente la misma idea. En Stern-Gerlach se usaba un campo magnético no uniforme y el momento magnético del espín, mientras que aquí usamos un campo eléctrico no uniforme y el momento dipolar eléctrico — el mecanismo físico es el mismo: "la energía depende de la intensidad del campo → en un campo no uniforme se experimenta una fuerza → se separa según el estado". Después de la separación, solo las moléculas en estado \(|I\rangle\) (alta energía) se envían a una cavidad resonante (cavity resonator).
Campo eléctrico dependiente del tiempo y transiciones¶
🟡 Lina: Dentro de la cavidad resonante existe un campo eléctrico que oscila con frecuencia \(\omega\): \(\mathcal{E}(t) = \mathcal{E}_0 \cos\omega t\). En este caso el hamiltoniano depende del tiempo, así que el enfoque de "encontrar autovalores y ya está" que hemos usado hasta ahora no funciona.
🔵 Kai: Un hamiltoniano dependiente del tiempo... eso parece difícil.
🟡 Lina: En general es difícil, pero cuando el campo es débil (\(\mu\mathcal{E}_0 \ll A\)) se puede resolver de forma aproximada. El truco es separar "la oscilación rápida sin campo" de "el cambio lento debido al campo".
🔵 Kai: ¿Separar? ¿Concretamente cómo se hace?
🟡 Lina: Sin campo eléctrico, la amplitud de un autoestado solo rota su fase rápidamente como \(C_I(t) = (\text{constante}) \times e^{-iE_I t/\hbar}\), ¿recuerdas? La idea es quitar de antemano esa "rotación rápida conocida" y seguir solo "el cambio nuevo que surge por el campo". Concretamente:
Sin campo eléctrico, \(\gamma_I\), \(\gamma_{II}\) son constantes que no cambian. Con campo, \(\gamma_I\), \(\gamma_{II}\) cambian lentamente. Así, en la ecuación de movimiento para \(\gamma\) desaparece la parte de "oscilación rápida" y solo queda el efecto del campo — lo que mejora mucho la visibilidad.
⚪ Mei: Entiendo, "restar la rotación conocida y ver solo el resto" — como eliminar el fondo.
🟡 Lina: Te mostraré el camino para reescribir la ecuación de movimiento en términos de \(\gamma_I\), \(\gamma_{II}\). Es un poco largo pero sigámoslo paso a paso.
Primero, derivando temporalmente \(C_I(t) = \gamma_I(t)\,e^{-iE_I t/\hbar}\) de (6.29), por la regla del producto:
Sustituyendo en el lado izquierdo \(i\hbar\,dC_I/dt\) de la ecuación (6.2):
Por otro lado, el lado derecho se escribe usando el hamiltoniano en presencia de campo \(H = H_0 + V(t)\). Aquí \(H_0\) es el hamiltoniano sin campo (con autovalores \(E_I\), \(E_{II}\)), y \(V(t)\) es la perturbación debida al campo. En la base de energía \(\{|I\rangle, |II\rangle\}\), la contribución de \(H_0\) es diagonal y genera \(E_I\,\gamma_I\,e^{-iE_I t/\hbar}\) (la parte correspondiente a la rotación estacionaria). Como el mismo término aparece en el lado izquierdo, se cancela en ambos lados, y solo quedan los términos no diagonales debidos a \(V(t)\).
🔵 Kai: Un momento. "Que \(V(t)\) sea no diagonal" — ¿en la base de posición \(\{|1\rangle, |2\rangle\}\) la perturbación \(V\) era diagonal, no? ¿Al cambiar a la base de energía se vuelve no diagonal?
🟡 Lina: Buena pregunta. Ese es exactamente el punto clave. En la base de posición, \(\langle 1|V|1\rangle = +\mu\mathcal{E}_0\cos\omega t\), \(\langle 2|V|2\rangle = -\mu\mathcal{E}_0\cos\omega t\), y \(\langle 1|V|2\rangle = \langle 2|V|1\rangle = 0\) — es decir, diagonal. Pero calculemos el elemento no diagonal \(\langle I|V|II\rangle\) en la base de energía. Sustituyendo \(|I\rangle = \frac{1}{\sqrt{2}}(|1\rangle - |2\rangle)\), \(|II\rangle = \frac{1}{\sqrt{2}}(|1\rangle + |2\rangle)\):
⚪ Mei: Ya veo. Una perturbación que era diagonal en la base de posición genera componentes no diagonales en la base de energía — se convierte en un término que conecta \(|I\rangle\) y \(|II\rangle\). Es interesante cómo al cambiar de base se intercambia lo "diagonal/no diagonal".
🟡 Lina: Exacto. Cambiar de base es como rotar los ejes de coordenadas — la apariencia de la matriz cambia, pero el contenido físico es el mismo. La derivación completa la puedes verificar en los ejercicios.
Bien, resumiendo: en el lado derecho, \(\gamma_{II}\) originalmente venía con la forma \(C_{II} = \gamma_{II}\,e^{-iE_{II} t/\hbar}\) y el factor \(e^{-iE_{II} t/\hbar}\), que al combinarse con el \(e^{+iE_I t/\hbar}\) que sale del lado izquierdo, produce un factor \(e^{-i(E_{II} - E_I)t/\hbar} = e^{i\omega_0 t}\) (donde \(\omega_0 = (E_I - E_{II})/\hbar = 2A/\hbar\)). Además, reescribiendo \(\cos\omega t = \frac{1}{2}(e^{i\omega t} + e^{-i\omega t})\), los factores que multiplican a \(\gamma_{II}\) son de dos tipos: \(e^{i(\omega_0 + \omega)t}\) y \(e^{i(\omega_0 - \omega)t}\). Es decir:
🔵 Kai: Aparecen dos tipos de factores oscilantes. \(\omega_0 + \omega\) y \(\omega_0 - \omega\)...
🟡 Lina: El primero (el término que oscila con \(\omega_0 + \omega\)) alterna rápidamente entre positivo y negativo a una frecuencia muy alta \(\omega_0 + \omega\). Por otro lado, ¿qué tan rápido cambia \(\gamma\)? El lado derecho completo de la ecuación es la "fuerza impulsora" que hace cambiar \(\gamma\), y su magnitud está determinada por el coeficiente \(\mu\mathcal{E}_0/(2\hbar)\). Recuerda la solución de \(dy/dt = ky\) que aprendiste en la preparatoria: \(y = y_0 e^{kt}\) — el tiempo que tarda \(y\) en cambiar significativamente es del orden de \(1/k\). Nuestra ecuación acoplada tiene la forma \(d\gamma_I/dt \sim (\mu\mathcal{E}_0/\hbar)\,\gamma_{II}\), así que el coeficiente \(\mu\mathcal{E}_0/\hbar\) del lado derecho determina "la velocidad del cambio". Estrictamente es un sistema acoplado y no es una exponencial simple, pero la escala temporal del cambio es del orden del inverso del coeficiente — es decir, \(\hbar/(\mu\mathcal{E}_0)\).
🔵 Kai: Es decir, ¿\(\gamma\) cambia lentamente? Pero, ¿por qué desaparece el "término que oscila rápidamente"? Aunque oscile rápido, no se hace cero...
🟡 Lina: Buena duda. Piénsalo así. Durante un breve intervalo de tiempo en que \(\gamma\) apenas cambia, el término de alta frecuencia \(e^{i(\omega_0+\omega)t}\) alterna entre positivo y negativo decenas de veces. La "fuerza" que hace cambiar \(\gamma\) se vuelve positiva y negativa a gran velocidad, así que lo empujado en dirección positiva y lo empujado en dirección negativa se cancelan casi completamente. Si empujas y jalas un columpio a altísima velocidad y de forma aleatoria, el columpio apenas se mueve, ¿verdad? Es lo mismo. En cambio, el término \(e^{i(\omega_0-\omega)t}\), cuando \(\omega \approx \omega_0\), casi no oscila y "empuja continuamente" en la misma dirección, así que su efecto se acumula.
⚪ Mei: La metáfora del columpio es muy clara. Una fuerza que alterna rápidamente entre positivo y negativo es efectivamente cero; solo la fuerza lenta tiene efecto.
🟡 Lina: Así es. Recuerda la condición \(\mu\mathcal{E}_0 \ll A\). Cuanto menor es el denominador, mayor es la fracción, así que si \(\mu\mathcal{E}_0 \ll A\) entonces \(\hbar/(\mu\mathcal{E}_0) \gg \hbar/A\) — es decir, la escala temporal del cambio de \(\gamma\), que es \(\hbar/(\mu\mathcal{E}_0)\), es mucho mayor que la escala temporal de la oscilación rápida \(\hbar/A\) (del orden del inverso de \(\omega_0 + \omega\)). Para dar una sensación numérica concreta: en el caso del amoníaco, \(\omega_0 = 2A/\hbar \sim 2\pi \times 24{,}000\) MHz, así que tomando \(\omega_0 + \omega \sim 2\omega_0\), un período de la oscilación rápida es del orden de \(10^{-11}\) segundos. Mientras tanto, si con un campo débil \(\mu\mathcal{E}_0\) es una centésima parte de \(A\), \(\gamma\) tarda del orden de \(10^{-9}\) segundos en cambiar significativamente — 100 veces más lento. Entonces el término de oscilación rápida alterna muchas veces entre positivo y negativo antes de que \(\gamma\) cambie apreciablemente, cancelándose y siendo efectivamente cero — esto se llama la aproximación de onda rotante (rotating wave approximation). El nombre proviene de que \(e^{i\omega t}\) representa una "onda que rota" en el plano complejo. Se descarta la componente que rota rápidamente y se deja solo la que rota lentamente — por eso "aproximación de onda rotante". Quedándonos solo con el segundo término, la ecuación para \(\gamma_I\) queda aproximadamente:
(Ecuaciones aproximadas bajo la aproximación de onda rotante. La derivación completa la puedes verificar en Problema M-2. Derivación de las oscilaciones de Rabi.) Como complemento sobre por qué el signo del exponente en (6.30b) es opuesto al de (6.30a): en la ecuación para \(\gamma_{II}\), al eliminar \(e^{-iE_{II} t/\hbar}\) se multiplica por \(e^{+iE_{II} t/\hbar}\), que al combinarse con el \(e^{-iE_I t/\hbar}\) que acompaña a \(\gamma_I\) produce \(e^{-i(E_I - E_{II})t/\hbar} = e^{-i\omega_0 t}\) — es decir, con el signo exactamente opuesto al de la ecuación de \(\gamma_I\). Las dos ecuaciones son simétricas y muestran que \(\gamma_I\) y \(\gamma_{II}\) están acopladas mutuamente.
🔵 Kai: La forma de la ecuación (6.30) se parece a la ecuación (6.2) del principio. \(\gamma_I\) y \(\gamma_{II}\) se influyen mutuamente.
🟡 Lina: El punto clave es el factor \(e^{\pm i(\omega_0 - \omega)t}\). Aquí \(\omega_0 = (E_I - E_{II})/\hbar = 2A/\hbar\) es la misma cantidad que la frecuencia angular de la oscilación de Rabi obtenida en (6.22). Cuando se cumple la condición de resonancia \(\omega \approx \omega_0\), \(e^{\pm i(\omega_0 - \omega)t} \approx 1\) permanece casi constante y el efecto se acumula, haciendo que la transición ocurra de la forma más eficiente.
🔵 Kai: ¿Resonancia...? ¿Por qué cuando las frecuencias coinciden la transición es más fácil?
🟡 Lina: Imagina un columpio. Si empujas al ritmo del período de oscilación, cada empujón se acumula y el columpio oscila cada vez más, ¿verdad? Pero si el ritmo no coincide, a veces empujas y a veces frenas, y los efectos se cancelan.
🔵 Kai: ¿Matemáticamente por qué ocurre esto?
🟡 Lina: Mira la ecuación (6.30) que acabamos de derivar. El factor \(e^{i(\omega_0 - \omega)t}\) del lado derecho es la clave. Volviendo a la metáfora del columpio, este factor representa "el desfase entre el ritmo del empujón y el ritmo de la oscilación". Si \(\omega = \omega_0\) entonces \(e^{i \cdot 0 \cdot t} = 1\), y siempre empujas al mismo ritmo — por eso el efecto se acumula en una sola dirección. Pero si \(\omega \neq \omega_0\), \(e^{i(\omega_0 - \omega)t}\) rota en el plano complejo: en ciertos momentos contribuye positivamente (empuja) y en otros negativamente (frena), y a largo plazo se cancela. Por eso, solo cuando la frecuencia del campo externo coincide exactamente con \(\omega_0 = 2A/\hbar\), la probabilidad de transición se acumula eficientemente. Cuando se cumple la condición de resonancia, la molécula transiciona de \(|I\rangle\) a \(|II\rangle\) y emite la diferencia de energía \(E_I - E_{II} = 2A\) como un fotón de microondas.
🔵 Kai: La diferencia de energía se convierte directamente en la energía del fotón. Por la relación \(E = h\nu\).
🟡 Lina: Esto es la emisión estimulada (stimulated emission) predicha por Einstein en 1917. El campo externo (las microondas ya presentes en la cavidad) "invita" a la molécula a emitir un fotón. El fotón emitido tiene la misma frecuencia y la misma fase que el campo externo, así que las microondas se amplifican.
⚪ Mei: Al tener la misma frecuencia y la misma fase, se obtiene una fuente de microondas coherente (con fase alineada).
🟡 Lina: Exacto. Resumiendo el funcionamiento del máser:
- Preparación de moléculas: Selección de moléculas en el estado de alta energía \(|I\rangle\) mediante campo eléctrico no uniforme
- Introducción en la cavidad resonante: Se envían las moléculas a una cavidad donde existen microondas con la frecuencia de resonancia \(\omega_0 = 2A/\hbar\)
- Emisión estimulada: Las moléculas transicionan \(|I\rangle \to |II\rangle\) y emiten fotones de microondas
- Amplificación: Los fotones emitidos refuerzan las microondas dentro de la cavidad
🟡 Lina: En Fig. 6.6「Principio de funcionamiento del máser de amoníaco」 he dibujado esquemáticamente estos 4 pasos.
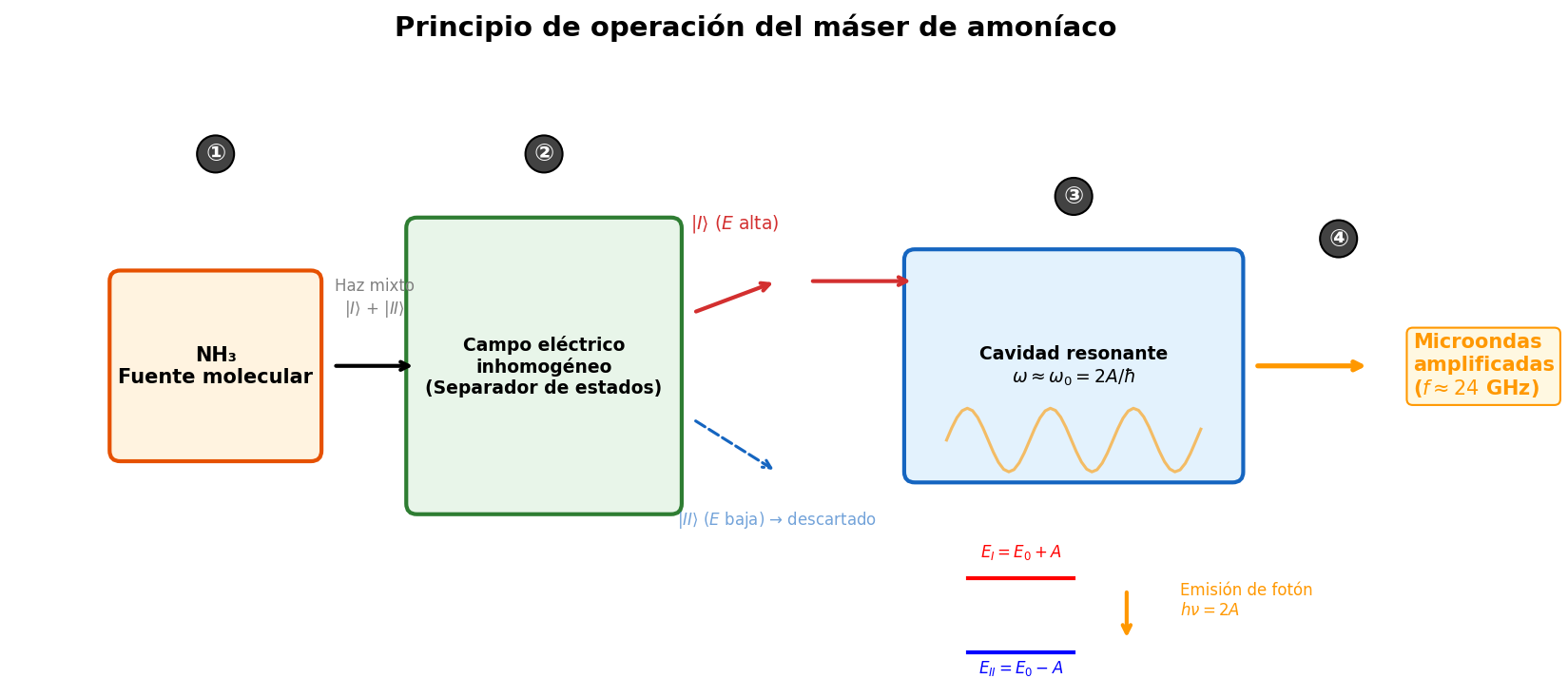
Fig. 6.6: Principio de funcionamiento del máser de amoníaco. ① Se emite un haz mixto desde la fuente de moléculas NH₃ → ② Se selecciona el estado de alta energía \(|I\rangle\) con campo eléctrico no uniforme → ③ Se introduce en la cavidad resonante con frecuencia de resonancia \(\omega_0 = 2A/\hbar\) provocando emisión estimulada → ④ Se obtiene como salida microondas coherentes amplificadas (aproximadamente 24 GHz).
🔵 Kai: ¡Increíble! La teoría del sistema de 2 estados de la mecánica cuántica se convierte directamente en el principio de funcionamiento de un dispositivo real... Pero tengo una duda. ¿Por qué hay que seleccionar solo las moléculas en estado de alta energía \(|I\rangle\)? ¿No valdría con las de baja energía \(|II\rangle\)?
🟡 Lina: Buena pregunta. Las moléculas en \(|II\rangle\) absorberían fotones y subirían a \(|I\rangle\) — es decir, debilitarían las microondas. Para amplificar, se necesita que haya más moléculas que emitan fotones. Por eso hay que seleccionar y enviar las de estado de alta energía. Esto se llama inversión de población (population inversion).
🔵 Kai: Entiendo que se necesitan más moléculas emitiendo. Pero, si simplemente se juntan moléculas, ¿no habría aproximadamente la mitad en alta energía y la mitad en baja? Sin separarlas, ¿no sería que emisión y absorción se equilibran y el resultado neto es cero?
🟡 Lina: Buen punto. En realidad, en condiciones normales (equilibrio térmico) hay más moléculas en el estado de baja energía \(|II\rangle\). A temperatura finita, las moléculas se concentran más en los estados de menor energía — este es un resultado fundamental de la mecánica estadística llamado distribución de Boltzmann. Así que sin hacer nada, las moléculas que absorben fotones (\(|II\rangle \to |I\rangle\)) son más numerosas que las que emiten (\(|I\rangle \to |II\rangle\)), y las microondas se debilitan. Para amplificar hay que hacer que el estado de alta energía sea artificialmente mayoritario — crear la inversión de población. Esta es la clave del máser.
🔵 Kai: Ya veo... naturalmente el estado de baja energía es el mayoritario, así que es necesario usar el campo no uniforme para seleccionar las moléculas de alta energía.
🟡 Lina: Exacto. Y extender este principio al dominio de la luz dio lugar al LASER (láser). Cuando tratemos cuantitativamente los coeficientes A y B de Einstein y la emisión estimulada en Cap. 21, volveremos a este tema.
✅ Verificación de comprensión: Explica por qué la condición de resonancia es importante en el máser de amoníaco.
Respuesta
Cuando la frecuencia del campo externo \(\omega\) coincide con la frecuencia \(\omega_0 = 2A/\hbar\) correspondiente a la diferencia entre los dos niveles de energía, la transición \(|I\rangle \to |II\rangle\) ocurre de la forma más eficiente. Si la frecuencia está desalineada, la probabilidad de transición disminuye drásticamente y no se produce la amplificación de microondas.
📝 Ejercicios:
- Hallar la longitud de onda del fotón correspondiente a la frecuencia de resonancia \(f_0 = \omega_0/(2\pi) = 24{,}000\) MHz de la molécula de amoníaco. ¿A qué región del espectro electromagnético pertenece? → Problema M-3. Niveles de energía de la molécula de amoníaco en un campo eléctrico
6.10 Fórmulas generales para el sistema de 2 estados — Resumen¶
🟡 Lina: Resumamos los resultados obtenidos en este capítulo para un hamiltoniano general de 2 estados
independiente del tiempo. Denotaremos los autovalores de energía como \(E_+\), \(E_-\).
🔵 Kai: Espera, hasta ahora escribíamos \(E_I\), \(E_{II}\), y ahora pasan a ser \(E_+\), \(E_-\). ¿Por qué el cambio de notación?
🟡 Lina: Buena observación. Como estamos resumiendo fórmulas generales no limitadas a la molécula de amoníaco, cambié la notación. \(E_I\), \(E_{II}\) eran etiquetas específicas del amoníaco, mientras que \(E_+\), \(E_-\) son símbolos generales "válidos para cualquier sistema de 2 estados". La correspondencia es simple: \(+\) es el más alto, \(-\) es el más bajo:
Para la molécula de amoníaco: \(E_+ = E_0 + A\), \(E_- = E_0 - A\). En adelante, para sistemas generales de 2 estados usaremos \(E_\pm\).
Autovalores de energía (ecuación (6.25) reformulada con la notación \(E_\pm\)):
Evolución temporal de los estados estacionarios:
Evolución temporal de un estado general:
donde \(c_{\pm} = \langle E_{\pm}|\psi(0)\rangle\) se determinan por la condición inicial.
⚪ Mei: Esta receta es un marco general aplicable a cualquier sistema de 2 estados, no solo a la molécula de amoníaco.
🟡 Lina: Así es. El sistema de 2 estados es el más básico en mecánica cuántica, pero una cantidad sorprendente de fenómenos físicos puede entenderse dentro de este marco. Por ejemplo, el comportamiento de una partícula de espín 1/2 en un campo magnético también. En Cap. 17 describiremos la partícula de espín 1/2 con las matrices de Pauli, pero matemáticamente es exactamente el hamiltoniano de 2 estados que hemos visto aquí.
🔵 Kai: Si resumo el contenido de hoy en una frase... "Si escribes la matriz hamiltoniana y resuelves el problema de autovalores, obtienes tanto los niveles de energía como la evolución temporal". Pero escribir correctamente el hamiltoniano parece ser lo más difícil. En el amoníaco la simetría determinó \(H_{11} = H_{22}\), y el efecto túnel permitió poner \(H_{12} = -A\). Pero si no hubiera simetría, o si hubiera 3 o más estados, ¿qué pistas se usarían para determinar el hamiltoniano?
🟡 Lina: Buen punto. De hecho, construir el hamiltoniano es en sí mismo un problema central de la física. Se usan diversas pistas: simetrías, datos experimentales, correspondencia con la mecánica clásica. Veremos muchos ejemplos concretos de ahora en adelante.
✅ Verificación de comprensión: En un sistema de 2 estados, ¿cómo cambia el desdoblamiento de energía \(E_+ - E_-\) entre el caso \(H_{11} = H_{22}\) (simétrico) y \(H_{11} \neq H_{22}\) (asimétrico)?
Respuesta
Caso simétrico: \(E_+ - E_- = 2|H_{12}|\). Caso asimétrico: \(E_+ - E_- = 2\sqrt{[(H_{11} - H_{22})/2]^2 + |H_{12}|^2}\). La asimetría \(H_{11} \neq H_{22}\) actúa en la dirección de aumentar el desdoblamiento de energía. El caso simétrico da el desdoblamiento mínimo.
📝 Ejercicios:
- Modelar el ion de hidrógeno molecular H₂⁺ como un sistema de 2 estados y expresar las energías del estado enlazante (baja energía) y antienlazante (alta energía) en términos de \(E_0\) y \(A\). → Problema M-4. Diagonalización del Hamiltoniano y transformación de la representación matricial
Adelanto del próximo capítulo¶
🟡 Lina: En este capítulo experimentamos el flujo "hamiltoniano → problema de autovalores → evolución temporal" en el escenario más simple: un sistema de 2 estados. Pero en la naturaleza hay muchos sistemas con infinitos estados.
🔵 Kai: ¡¿Infinitos?! ¿Eso significa que la matriz se vuelve infinitamente grande?
🟡 Lina: Así es. Por ejemplo, una partícula moviéndose en una línea recta unidimensional puede estar en principio en cualquier posición, así que el número de estados base es continuo e infinito. En ese caso, la amplitud \(C_i\) ya no es "una función del índice \(i\)", sino "una función de la posición \(x\)" — es decir, la función de onda \(\psi(x, t)\).
⚪ Mei: Si la matriz se vuelve infinitamente grande, ya no se puede manejar con cálculos matriciales ordinarios, ¿verdad...?
🟡 Lina: Así es. De hecho, la "matriz" hamiltoniana en el caso continuo se convierte en un operador diferencial. En el próximo Cap. 7, extenderemos el formalismo matricial de este capítulo al caso continuo e introduciremos la ecuación de Schrödinger. Confirmaremos que la intuición cultivada con el sistema de 2 estados — "el hamiltoniano determina la energía, los autoestados son los estados estacionarios, y un estado general es una superposición de autoestados" — se puede usar tal cual.
🔵 Kai: ¡Estoy deseando verlo!
Referencias¶
- R. P. Feynman, R. B. Leighton, M. Sands, The Feynman Lectures on Physics, Vol. III (Addison-Wesley). Capítulo 8 "The Hamiltonian Matrix", Capítulo 9 "The Ammonia Maser". La formulación del sistema de 2 estados y la introducción de la matriz hamiltoniana de este capítulo siguen el enfoque pedagógico de Feynman.
- J. J. Sakurai, J. Napolitano, Modern Quantum Mechanics, 3ª edición (Cambridge University Press). El Capítulo 2 "Quantum Dynamics" introduce axiomáticamente el operador de evolución temporal y la ecuación de Schrödinger, proporcionando las bases matemáticas del enfoque axiomático de este capítulo.
- A. Shimizu, Fundamentos de la teoría cuántica — Para una comprensión sencilla de su esencia (Saiensu-sha). Explicación cuidadosa de la estructura básica de la mecánica cuántica utilizando sistemas de 2 estados.
- K. Hiroe, Mecánica cuántica como hobby. Explicación accesible del sistema de 2 estados y las oscilaciones de Rabi.
Feedback on this page
Let us know if something was unclear, incorrect, or could be improved.