Capítulo 3 Querer mejorar la eficiencia de la máquina de vapor — El nacimiento de la termodinámica y la entropía¶
Resumen de los capítulos anteriores: En Cap. 1 Newton creó un modelo de la gravedad, y en Cap. 2 Maxwell unificó la electricidad y el magnetismo. En ambos casos, la "curiosidad" fue la motivación principal. En este capítulo, la motivación es completamente diferente: veremos la historia de un modelo nacido de una necesidad práctica. Y veremos cómo una pregunta práctica sobre la eficiencia de las máquinas de vapor alcanza una propiedad fundamental del universo: la entropía.
Objetivo de este capítulo
- Seguir la historia de cómo una búsqueda iniciada por "necesidad" alcanzó una propiedad fundamental del universo
- Comprender el límite de eficiencia de Carnot y la entropía de Boltzmann, y captar la esencia de la mecánica estadística: que "contar los estados del mundo microscópico" explica los fenómenos térmicos macroscópicos
3.1 Motivación: ¿Se puede aumentar más la eficiencia de la máquina de vapor?¶
🟡 Lina: En Cap. 1 y Cap. 2 hablamos de cómo la curiosidad —"¿por qué se mueven los planetas?", "¿se pueden unificar la electricidad y el magnetismo?"— generó modelos. Hoy la motivación es completamente distinta: el dinero.
🔵 Kai: ¿El dinero?
🟡 Lina: Inglaterra en el siglo XVIII. En plena Revolución Industrial. Se quemaba carbón para producir vapor, y la fuerza del vapor movía las máquinas: la máquina de vapor era el corazón de la economía. Pero la eficiencia de las máquinas de vapor era baja. De toda la energía térmica introducida, solo una pequeña parte se convertía en trabajo útil. El resto se desechaba como calor residual.
🔵 Kai: Eso es un desperdicio. ¿Cuánto se perdía?
🟡 Lina: Las primeras máquinas de vapor tenían una eficiencia de apenas unos pocos por ciento: más del 90% del calor introducido se desperdiciaba. Por eso la pregunta "¿se puede aumentar más la eficiencia? ¿Existe un límite teórico?" era urgente. Quien respondió fue el francés Sadi Carnot. En su artículo de 1824, Reflexiones sobre la potencia motriz del fuego.
🔭 Nota de filosofía de la ciencia: Los modelos físicos nacen no solo de la "curiosidad" sino también de la "necesidad práctica". Sin embargo, sin importar cuál sea la motivación inicial, se puede llegar a lugares profundos. El tipo de motivación no determina el valor del modelo.
✅ Verificación de comprensión: ¿Quién fue el primero en mostrar el límite teórico de eficiencia de las máquinas de vapor?
Respuesta
Sadi Carnot. Lo mostró en su artículo de 1824, Reflexiones sobre la potencia motriz del fuego.
✅ Verificación de comprensión: ¿En qué se diferencia la motivación del modelo de este capítulo respecto a Cap. 1 y Cap. 2?
Respuesta
En Cap. 1 y Cap. 2 la motivación fue la curiosidad, pero en este capítulo es la necesidad práctica (el dinero) de mejorar la eficiencia de las máquinas de vapor.
3.2 La pregunta de Carnot — ¿Existe un límite para la eficiencia?¶
🟡 Lina: La pregunta de Carnot era simple. ¿Existe un límite teórico superior para la eficiencia de convertir calor en trabajo?
🔵 Kai: ¿Hay un límite superior? Parece que con el avance de la tecnología se podría aumentar la eficiencia sin fin.
🟡 Lina: Pues resulta que sí hay un límite superior. Hay una barrera que no se puede superar sin importar cuánto avance la tecnología. Carnot lo dedujo no mediante experimentos, sino mediante un experimento mental. Primero, organicemos qué hace una máquina térmica. Mira la Fig. 3.1「Flujo de energía de una máquina térmica」. En la figura, la temperatura de la fuente caliente se escribe como \(T_\text{hot}\) y la de la fuente fría como \(T_\text{cold}\), para explicitar "caliente (hot)" y "frío (cold)". De aquí en adelante en el texto usaré la notación abreviada \(T_H\) (\(H\) = hot) y \(T_C\) (\(C\) = cold). De manera similar, el calor se escribe como \(Q_H\) (calor recibido de la fuente caliente) y \(Q_C\) (calor cedido a la fuente fría).
%%{init: {"theme": "default", "themeCSS": ".edgePath .path, .flowchart-link { stroke-width: 2px !important; }"}}%%
flowchart LR
Hot["Fuente caliente<br/>Temperatura T_hot"] -->|"Calor Q_hot"| Engine["Máquina térmica"]
Engine -->|"Trabajo W"| Work["Trabajo<br/>(salida útil)"]
Engine -->|"Calor residual Q_cold"| Cold["Fuente fría<br/>Temperatura T_cold"]
style Hot fill:#f66,color:#fff
style Cold fill:#66f,color:#fff
style Engine fill:#ff9Fig. 3.1: Flujo de energía de una máquina térmica
Una máquina térmica recibe calor \(Q_H = Q_\text{hot}\) de la fuente caliente (temperatura \(T_H = T_\text{hot}\)), convierte una parte en trabajo \(W\), y cede el resto \(Q_C = Q_\text{cold}\) a la fuente fría (temperatura \(T_C = T_\text{cold}\)).
Los 4 pasos del ciclo de Carnot¶
🟡 Lina: Carnot pensó: "¿Y si existiera una máquina térmica ideal perfectamente sin desperdicios?" Construyó un ciclo ideal donde todos los procesos son reversibles (se pueden revertir). Consta de 4 pasos.
- Expansión isotérmica (a temperatura \(T_H\)): "Isotérmico" significa a temperatura constante. Se expande el gas manteniéndolo en contacto con la fuente caliente. Para que la temperatura no cambie, absorbe calor \(Q_H\) de la fuente mientras realiza trabajo.
- Expansión adiabática: "Adiabático" significa sin intercambio de calor. Se desconecta de la fuente caliente y se sigue expandiendo. Sin intercambio de calor, la temperatura baja de \(T_H\) a \(T_C\).
- Compresión isotérmica (a temperatura \(T_C\)): Se comprime el gas manteniéndolo en contacto con la fuente fría. Para que la temperatura no cambie, el gas cede calor \(Q_C\) a la fuente fría.
- Compresión adiabática: Se desconecta de la fuente fría y se sigue comprimiendo. Sin intercambio de calor, la temperatura sube de \(T_C\) a \(T_H\).
⚪ Mei: Subir y bajar la temperatura con procesos "adiabáticos", intercambiar calor con procesos "isotérmicos" — los roles están claramente separados.
🟡 Lina: Así es. He dibujado lo que ocurre físicamente en cada paso (Fig. 3.2「Los 4 pasos del ciclo de Carnot」).
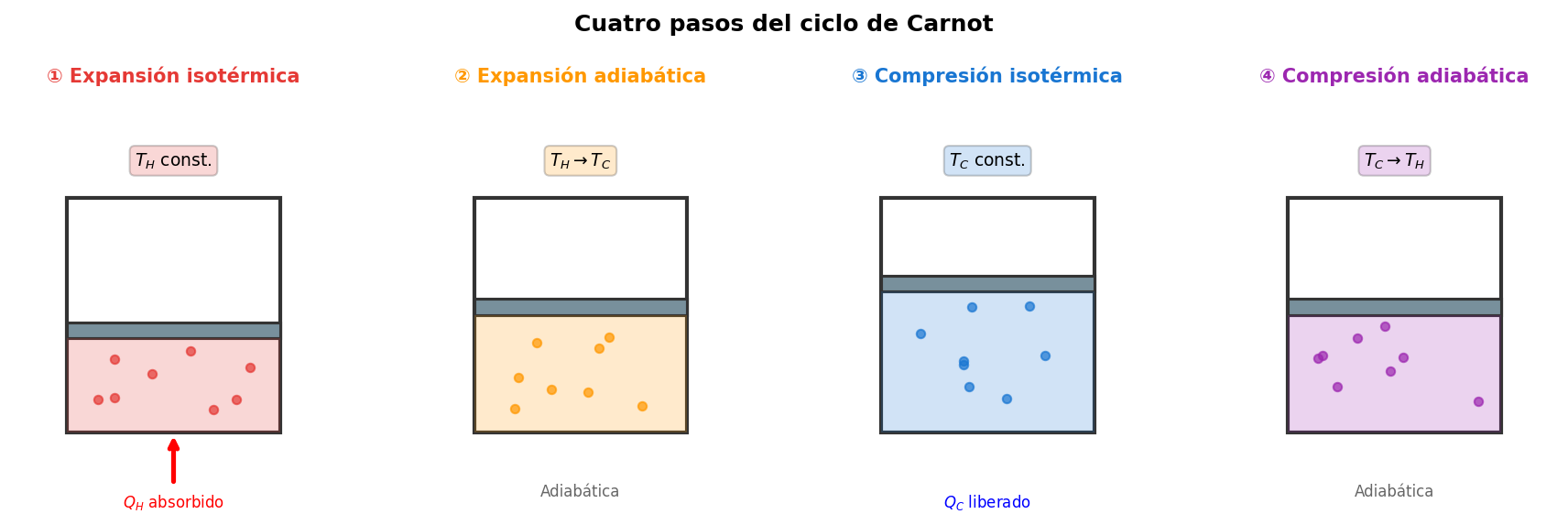
Fig. 3.2: Los 4 pasos del ciclo de Carnot. Ilustración física de cada paso con cilindro y pistón. ① Absorción de calor \(Q_H\) de la fuente caliente y expansión, ② expansión adiabática con descenso de temperatura, ③ cesión de calor \(Q_C\) a la fuente fría y compresión, ④ compresión adiabática con recuperación de temperatura.
🟡 Lina: Si dibujamos estos 4 pasos en un diagrama presión-volumen (diagrama PV), se ve como en Fig. 3.3「Diagrama PV del ciclo de Carnot」.
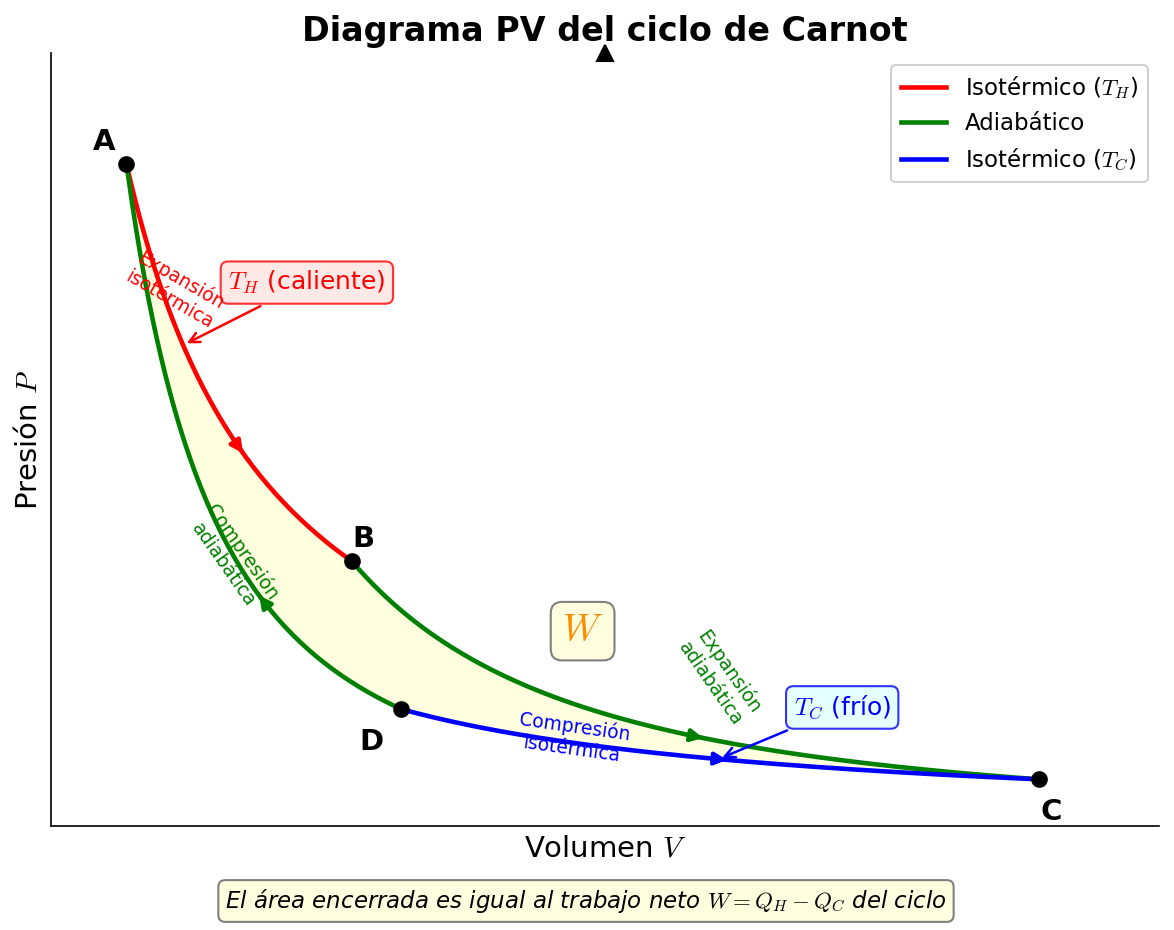
Fig. 3.3: Diagrama PV del ciclo de Carnot. Muestra los 4 pasos del ciclo de Carnot (expansión isotérmica → expansión adiabática → compresión isotérmica → compresión adiabática) en un diagrama presión-volumen. El área encerrada corresponde al trabajo neto \(W\).
🟡 Lina: Tras los 4 pasos, el gas vuelve a su estado original — por eso se llama "ciclo".
🔵 Kai: Si vuelve al estado original... ¿la energía interna también vuelve al original?
🟡 Lina: Exacto. Si el estado vuelve al original, el cambio en la energía interna es cero. Por conservación de la energía:
Es decir, el trabajo es la diferencia entre "el calor absorbido" y "el calor cedido".
🔵 Kai: Entiendo, solo la diferencia neta se convierte en trabajo. Pero si pudiéramos hacer \(Q_C\) igual a cero, ¿todo se convertiría en trabajo...?
Definición de eficiencia y conclusión de Carnot¶
🟡 Lina: La eficiencia \(\eta\) se define como "qué fracción del calor introducido se convierte en trabajo":
🔵 Kai: ¿Para obtener eficiencia del 100% basta con \(Q_C = 0\), es decir, cero calor residual? Pero, ¿hay alguna razón física por la que no se pueda eliminar el calor residual?
🟡 Lina: Ese es exactamente el punto central. Carnot mostró que en un ciclo reversible se cumple \(Q_H/T_H = Q_C/T_C\) — es decir, si \(T_C > 0\) entonces necesariamente \(Q_C > 0\), y no se puede eliminar el calor residual.
🔵 Kai: Eh, ¿por qué se cumple \(Q_H/T_H = Q_C/T_C\)? Que al dividir por la temperatura resulten iguales no me resulta intuitivo.
🟡 Lina: Buena pregunta. Ahora voy a deducirlo concretamente.
Derivación de la eficiencia de Carnot¶
🟡 Lina: Usando un gas ideal como sustancia de trabajo, podemos calcular cada paso concretamente. La ecuación de estado del gas ideal es \(pV = Nk_B T\). En el instituto quizás aprendiste \(pV = nRT\) (\(n\) es la cantidad de sustancia, \(R\) la constante de los gases), pero reescrita para \(N\) moléculas da \(Nk_B = nR\), así que es la misma ecuación. Aquí \(k_B \approx 1.381 \times 10^{-23}\;\text{J/K}\) es la constante de Boltzmann — un factor de conversión que conecta energía y temperatura a nivel de una sola molécula. Su relación con la constante de los gases \(R\) del instituto es \(R = N_A k_B\) (\(N_A\) es el número de Avogadro). En este capítulo escribiremos usando el número de moléculas \(N\) porque es más fácil conectar con la mecánica estadística más adelante.
Paso 1 (Expansión isotérmica \(A \to B\), temperatura \(T_H\)):
Primero, la energía interna es la suma total de la energía que poseen todas las partículas del sistema — la energía cinética de las partículas más la energía potencial de interacción entre ellas. En un gas ideal no hay fuerzas entre partículas, así que la energía potencial es cero y la energía interna es toda cinética — depende solo de la rapidez de las partículas. En un proceso isotérmico la temperatura es constante, así que la rapidez promedio de las partículas también es constante — por tanto la energía interna no cambia.
🔵 Kai: Entiendo, si la temperatura no cambia, el movimiento de las partículas tampoco cambia, así que la energía interna permanece constante.
🟡 Lina: Exacto. Ahora usemos la conservación de la energía. El calor añadido al sistema se reparte entre el aumento de energía interna y el trabajo realizado por el sistema — es decir, "calor añadido = cambio de energía interna + trabajo realizado". En forma de ecuación:
Aquí \(dU\) es el cambio infinitesimal de energía interna, \(\delta Q\) es la cantidad infinitesimal de calor añadido, y \(p\,dV\) es el trabajo infinitesimal realizado por el sistema cuando el volumen cambia en \(dV\). Quizás te llame la atención que uso símbolos diferentes \(\delta\) y \(d\), pero por ahora piensa que "ambos representan cantidades infinitesimales". La razón de la diferencia la explicaré en 「Primera ley (Conservación de la energía)」. Esto es la conservación de la energía en sí, y en una sección posterior le daremos el nombre formal de primera ley.
Por ahora recuerda "calor añadido = cambio de energía interna + trabajo realizado". En un proceso isotérmico el cambio de energía interna es cero, así que \(\delta Q = p\,dV\). Es decir, el calor absorbido es la suma (integral) de \(\delta Q = p\,dV\) a lo largo de todo el proceso. Sustituyendo \(p = Nk_BT_H/V\) de la ecuación de estado \(pV = Nk_BT_H\):
Aquí usamos \(\int_{V_A}^{V_B} \frac{dV}{V} = [\ln V]_{V_A}^{V_B} = \ln V_B - \ln V_A = \ln\frac{V_B}{V_A}\). Ya aprendiste en el instituto que \(\int 1/x\,dx = \ln x\).
⚪ Mei: El calor absorbido está determinado por la temperatura y el logaritmo de la razón de volúmenes. Cuanto más se expande el volumen, más calor absorbe.
Paso 3 (Compresión isotérmica \(C \to D\), temperatura \(T_C\)):
🟡 Lina: Como es un proceso isotérmico, al igual que en el paso 1, el cambio de energía interna es cero y se cumple \(\delta Q = p\,dV\). Pero esta vez la temperatura es \(T_C\), así que la ecuación de estado da \(p = Nk_BT_C/V\). En el paso 1 había expansión (\(dV > 0\)) así que \(\delta Q > 0\) — el sistema absorbía calor. Ahora es una compresión, así que \(dV < 0\) — es decir \(p\,dV < 0\), y \(\delta Q = p\,dV < 0\). Como \(\delta Q\) es "calor añadido al sistema", que sea negativo significa "el calor salió del sistema" — es decir, el sistema está cediendo calor. El calor total añadido al sistema durante todo el proceso se obtiene integrando mientras el volumen cambia de \(V_C\) a \(V_D\) (como es compresión, \(V_D < V_C\)):
\(\int_{V_C}^{V_D} \frac{dV}{V} = [\ln V]_{V_C}^{V_D} = \ln V_D - \ln V_C = \ln(V_D/V_C)\) se calcula mecánicamente y da el resultado correcto.
Como \(V_D < V_C\) (porque es compresión y el volumen disminuye), \(\ln(V_D/V_C) < 0\) — es decir, el resultado de la integral \(Nk_B T_C \ln(V_D/V_C)\) es un valor negativo. Efectivamente el sistema está cediendo calor.
Aquí queremos definir \(Q_C\) como "la magnitud del calor cedido por el sistema a la fuente fría", un valor positivo. Como el resultado de la integral es negativo (indica que "el sistema perdió calor"), basta tomar su valor absoluto. Es decir:
La última igualdad usa \(-\ln(a/b) = \ln(b/a)\).
🔵 Kai: ¿Y cómo se usan los dos procesos adiabáticos?
🟡 Lina: En los procesos adiabáticos \(\delta Q = 0\), así que la primera ley queda \(dU = -p\,dV\). Para un gas ideal monoatómico (un gas de un solo átomo como el helio), las partículas solo se mueven en las 3 direcciones \(x, y, z\), y se sabe experimentalmente que tienen una energía de \(\frac{1}{2}k_BT\) por cada dirección (\(k_B\) es la constante de Boltzmann que ya apareció en la ecuación de estado). Es decir, \(\frac{3}{2}k_BT\) por partícula, y para \(N\) partículas \(U = \frac{3}{2}Nk_BT\). Por qué se cumple esto lo derivaremos de la mecánica estadística en la segunda mitad de este capítulo (3.7「Significado estadístico de la temperatura」). Por ahora úsalo como un hecho experimental. El cambio infinitesimal es \(dU = \frac{3}{2} N k_B\,dT\). Combinando esto con la ecuación de estado \(p = Nk_BT/V\), derivemos cómo se relacionan temperatura y volumen en un proceso adiabático. Sustituyendo en \(dU = -p\,dV\):
Dividiendo ambos lados por \(Nk_B T\) obtenemos \(\frac{3}{2}\frac{dT}{T} = -\frac{dV}{V}\). Observa que "el lado izquierdo es una expresión solo en \(T\), el lado derecho solo en \(V\)". En estos casos se puede integrar cada lado respecto a su propia variable (método llamado separación de variables). Usando \(\int dT/T = \ln T\) e \(\int dV/V = \ln V\), obtenemos \(\frac{3}{2}\ln T = -\ln V + \text{const}\). Pasando \(\ln V\) al lado izquierdo: \(\frac{3}{2}\ln T + \ln V = \text{const}\). Usando las propiedades del logaritmo \(a\ln x = \ln x^a\) y \(\ln x + \ln y = \ln(xy)\): \(\ln(T^{3/2} V) = \text{const}\). Tomando la exponencial de ambos lados: \(T^{3/2} V = \text{constante}\).
🔵 Kai: Oh, la temperatura y el volumen están determinados conjuntamente. Si se expande, la temperatura baja.
🟡 Lina: Así es. Esto se puede usar tal cual, pero para hacer más visible "la relación entre \(T\) y \(V\)" quiero que el exponente de \(T\) sea 1, así que elevo ambos lados a la potencia \(2/3\). \((T^{3/2} V)^{2/3} = T^{(3/2)(2/3)} \cdot V^{2/3} = T \cdot V^{2/3}\), así que \(T V^{2/3} = \text{constante}\) (el valor concreto de la constante cambia pero sigue siendo "constante"). Quizás en el instituto aprendiste \(pV^\gamma = \text{constante}\) (\(\gamma\) es la razón de capacidades caloríficas); para un gas ideal monoatómico \(\gamma = 5/3\), y \(TV^{\gamma-1} = TV^{2/3} = \text{constante}\) es la misma ecuación. Lo importante es que "en un proceso adiabático, temperatura y volumen no pueden variar independientemente". Aplicando esto a la expansión adiabática \(B \to C\) y la compresión adiabática \(D \to A\):
Dividamos la primera ecuación por la segunda. El lado izquierdo es \(\frac{T_H V_B^{2/3}}{T_C V_D^{2/3}}\), el derecho \(\frac{T_C V_C^{2/3}}{T_H V_A^{2/3}}\) — ah, se complica un poco. Hagámoslo de forma más sencilla. De la primera ecuación: \(V_B^{2/3} = (T_C/T_H) V_C^{2/3}\); de la segunda: \(V_A^{2/3} = (T_C/T_H) V_D^{2/3}\). Dividiendo estas dos, \(T_C/T_H\) se cancela:
Eliminando la potencia \(2/3\) (elevando ambos lados a \(3/2\)):
🔵 Kai: ¡Ah, el argumento de los logaritmos es el mismo! Entonces si tomamos la razón \(Q_C/Q_H\)... ¿los logaritmos se cancelan?
🟡 Lina: Exacto. Como \(\ln(V_B/V_A) = \ln(V_C/V_D)\), al tomar la razón de \(Q_H\) y \(Q_C\) los factores logarítmicos se cancelan y solo queda la razón de temperaturas:
Por tanto, la eficiencia del ciclo de Carnot es:
🔵 Kai: Esto se calculó con un gas ideal monoatómico, ¿verdad? ¿Cambiaría con otro gas?
🟡 Lina: Buena pregunta. En realidad, la eficiencia de Carnot no depende de la sustancia de trabajo. Aquí calculamos concretamente con un gas ideal monoatómico, pero ya sea una molécula diatómica o un líquido, si el ciclo es reversible se obtiene el mismo resultado. Esto es consecuencia del teorema de Carnot: si la eficiencia dependiera de la sustancia de trabajo, se podría combinar una de alta eficiencia con una de baja eficiencia para construir un dispositivo que contradiga la segunda ley.
⚪ Mei: Es decir, mientras la temperatura de la fuente fría no sea cero, la eficiencia no puede ser del 100%.
🟡 Lina: Además, lo importante es el teorema de Carnot: entre todas las máquinas térmicas que operan entre las mismas temperaturas, la máquina reversible (máquina de Carnot) tiene la eficiencia máxima. Las máquinas irreversibles necesariamente tienen menor eficiencia.
🔵 Kai: ¿Por qué?
🟡 Lina: Si existiera una máquina con mayor eficiencia que la de Carnot, combinándola con la máquina de Carnot funcionando al revés (bomba de calor), sería posible "transferir calor de la fuente fría a la caliente sin ningún costo". Esto contradice la segunda ley que veremos más adelante.
📝 Ejercicios:
- Cálculo concreto de la eficiencia de Carnot → Problema B-1. Cálculo de la eficiencia de Carnot
✅ Verificación de comprensión: Escribe la fórmula que expresa el límite superior de eficiencia del ciclo de Carnot.
Respuesta
\(\eta_{\text{Carnot}} = 1 - \frac{T_C}{T_H}\)
✅ Verificación de comprensión: ¿En qué caso el límite de eficiencia de Carnot no alcanza el 100%?
Respuesta
Mientras la temperatura de la fuente fría \(T_C\) no sea cero, la eficiencia no puede ser del 100%.
3.3 Las leyes de la termodinámica — Conservación de la energía y dirección¶
🟡 Lina: A partir del trabajo de Carnot, en el siglo XIX se establecieron las leyes de la termodinámica. Hay desde la ley cero hasta la tercera ley, pero las más importantes son la ley cero, la primera ley y la segunda ley.
Ley cero (Transitividad del equilibrio térmico)¶
🟡 Lina: La ley cero es la ley que garantiza la existencia de la temperatura.
Si el sistema \(A\) está en equilibrio térmico con el sistema \(C\), y el sistema \(B\) también está en equilibrio térmico con el sistema \(C\), entonces \(A\) y \(B\) están en equilibrio térmico entre sí.
⚪ Mei: Suena obvio...
🟡 Lina: Parece "obvio", pero es precisamente porque esta transitividad se cumple que podemos etiquetar todos los sistemas con un solo número llamado "temperatura". Si esta ley no se cumpliera, podría ocurrir que "\(A\) y \(C\) están en equilibrio pero \(A\) y \(B\) no", y el concepto mismo de temperatura perdería su sentido.
✅ Verificación de comprensión: ¿Por qué es importante la ley cero de la termodinámica? ¿Qué problema habría si no se cumpliera?
Respuesta
Precisamente porque se cumple la ley cero (transitividad del equilibrio térmico), podemos etiquetar todos los sistemas con un solo número llamado "temperatura". Si no se cumpliera, el concepto mismo de temperatura perdería su sentido.
Primera ley (Conservación de la energía)¶
🟡 Lina: La primera ley es la conservación de la energía. En forma de cambios infinitesimales:
Donde \(dU\) es el cambio de energía interna, \(\delta Q\) es el calor añadido al sistema, y \(\delta W\) es el trabajo realizado por el sistema. Si consideramos expansión/compresión de un gas, \(\delta W = p\,dV\), así que:
Esto es exactamente la misma ecuación que usamos en la derivación del ciclo de Carnot —"calor añadido = cambio de energía interna + trabajo realizado" (\(\delta Q = dU + p\,dV\))— resuelta para \(dU\).
🔵 Kai: ¿Por qué \(d\) y \(\delta\) son diferentes?
🟡 Lina: Buena pregunta. \(dU\) es un cambio infinitesimal de una variable de estado — si el estado del sistema está determinado, el valor de \(U\) queda determinado de manera única. En cambio, \(\delta Q\) y \(\delta W\) son cantidades que dependen del camino — su valor cambia según qué proceso se siga. Es la misma diferencia que entre el saldo de una cuenta bancaria (variable de estado) y la cantidad de un depósito en efectivo (depende del camino).
⚪ Mei: Organizándolo queda así.
Tabla 3.1: Comparación entre variables de estado y cantidades dependientes del camino
| Clasificación | Símbolo | Ejemplo | Característica |
|---|---|---|---|
| Variable de estado (\(d\)) | \(dU\), \(dS\), \(dV\) | Energía interna, entropía, volumen | El valor queda determinado de manera única por el estado. No depende del camino |
| Cantidad dependiente del camino (\(\delta\)) | \(\delta Q\), \(\delta W\) | Calor, trabajo | El valor cambia según qué proceso se siga |
✅ Verificación de comprensión: En la ecuación de la primera ley \(dU = \delta Q - \delta W\), ¿por qué se usan los símbolos \(d\) y \(\delta\) de forma diferente?
Respuesta
\(dU\) representa el cambio infinitesimal de una variable de estado (cuyo valor queda determinado de manera única por el estado del sistema), mientras que \(\delta Q\) y \(\delta W\) representan cantidades que dependen del camino (cuyo valor cambia según qué proceso se siga).
Segunda ley (Aumento de la entropía · Irreversibilidad)¶
🟡 Lina: La segunda ley es la ley de la "dirección". Tiene varias formulaciones equivalentes:
Formulación de Clausius: El calor no fluye espontáneamente de un cuerpo frío a uno caliente.
Formulación de Kelvin: No existe ningún proceso cuyo único resultado sea extraer calor de una fuente térmica y convertirlo completamente en trabajo.
🔵 Kai: ¿Eso no es obvio? El café caliente se enfría, pero el café frío nunca se calienta solo.
🟡 Lina: Parece "obvio", ¿verdad? Pero en realidad es un misterio profundo. La ecuación de Newton \(F = ma\) es simétrica bajo inversión temporal — es decir, si inviertes el tiempo la forma de la ecuación no cambia. El movimiento microscópico de las partículas no contradice las leyes físicas ni hacia adelante ni hacia atrás en el tiempo.
🔵 Kai: Eh, espera un momento. Si las leyes microscópicas son simétricas bajo inversión temporal... ¿eso significa que microscópicamente está permitido que "el calor fluya de frío a caliente"? Pero ¿eso no es una contradicción? Si microscópicamente está bien pero macroscópicamente nunca ocurre. Entonces, ¿la segunda ley no es una "ley absoluta" como las leyes de Newton?
🟡 Lina: Buena intuición. De hecho es así — el carácter de la segunda ley es esencialmente diferente de la ecuación del movimiento de Newton. Quien resolvió esta contradicción fue Boltzmann — pero antes, veamos la definición termodinámica de la entropía.
✅ Verificación de comprensión: Escribe la primera ley de la termodinámica en forma de cambios infinitesimales.
Respuesta
\(dU = \delta Q - p\,dV\) (el calor añadido al sistema es igual, por conservación de la energía, a la suma del cambio de energía interna y el trabajo realizado por el sistema).
✅ Verificación de comprensión: ¿Cuál es la formulación de Clausius de la segunda ley de la termodinámica?
Respuesta
El calor no fluye espontáneamente de un cuerpo frío a uno caliente.
3.4 Definición termodinámica de la entropía¶
🟡 Lina: Del resultado del ciclo de Carnot se puede deducir algo sorprendente. En un ciclo reversible:
Déjame reorganizar un poco la notación. Hasta ahora tratamos tanto \(Q_H\) como \(Q_C\) como valores positivos que representan "magnitudes de calor". Pero a partir de ahora, para generalizar, cambiaré al convenio de signos positivo para calor recibido por el sistema, negativo para calor cedido por el sistema. Con este convenio, el calor cedido por el sistema en la compresión isotérmica se escribe como \(-Q_C\). Entonces la ecuación anterior se convierte en:
🔵 Kai: Si sumamos al dar una vuelta completa da cero... ¿es coincidencia?
🟡 Lina: No es coincidencia. De hecho, esto se puede generalizar. El ciclo de Carnot es una curva cerrada de forma especial, pero quiero mostrar que lo mismo se cumple para cualquier ciclo reversible de forma arbitraria. Cuando se dibuja una curva cerrada arbitraria en el diagrama PV, consideremos dividir su interior en una rejilla fina. La razón por la que se puede construir una rejilla es que en cada punto del diagrama PV se puede trazar "una curva de temperatura constante (isoterma)" y "una curva sin intercambio de calor (adiabática)" que pasen por ese punto. Las isotermas son "el conjunto de \((p, V)\) que satisfacen la ecuación de estado \(pV = Nk_BT\) para \(T\) fijo", así que existe una para cada temperatura. Las adiabáticas también existen como curvas que satisfacen "\(TV^{2/3} = \text{constante}\)" pasando una por cada punto. Es exactamente como en un mapa, donde por cualquier punto pasan un "meridiano" y un "paralelo".
🔵 Kai: Ah, como meridianos y paralelos en un mapa, en el diagrama PV las isotermas y adiabáticas se cruzan en todas partes formando una rejilla.
🟡 Lina: Exacto. Usando isotermas y adiabáticas como líneas de rejilla, podemos dividir el diagrama PV en celdas finas. Cada celda tiene 4 lados "isotérmico → adiabático → isotérmico → adiabático" — es decir, es un ciclo de Carnot infinitesimal. La relación \(Q_H/T_H = Q_C/T_C\) que mostramos para el gas ideal es, de hecho, independiente de la sustancia de trabajo por el teorema de Carnot — para cualquier sustancia, en un ciclo de Carnot reversible se cumple la misma relación (si hubiera una sustancia que no la cumpliera, combinándola con un gas ideal se podría construir un dispositivo que contradiga la segunda ley). Por tanto, para cada ciclo de Carnot infinitesimal también se cumple \(\delta Q_H/T_H + \delta Q_C/T_C = 0\). Si llenamos el interior de cualquier curva cerrada con estas celdas, podemos aproximar la curva como una colección de ciclos de Carnot infinitesimales — como aproximar una curva con forma de escalera.
🔵 Kai: Lo de que los lados compartidos entre ciclos adyacentes se cancelan me cuesta un poco visualizarlo...
🟡 Lina: Piénsalo así. Imagina alicatar un suelo. Cuando cubres todo el suelo con pequeños azulejos cuadrados, la línea de borde entre dos azulejos adyacentes es compartida por ambos. Desde la perspectiva de un azulejo es "el lado derecho", pero desde el azulejo vecino es "el lado izquierdo" — se recorre el mismo lado en dirección opuesta.
Físicamente hablando, el lado de expansión isotérmica de un ciclo infinitesimal es, visto desde el ciclo infinitesimal vecino, un lado de compresión isotérmica. Se recorre el mismo tramo de la misma isoterma, uno en dirección de expansión (\(\delta Q > 0\)) y el otro en dirección de compresión (\(\delta Q < 0\)). Como la temperatura es la misma y la magnitud del cambio es la misma, las contribuciones de \(\delta Q/T\) se cancelan exactamente.
🔵 Kai: Ah, es decir, al sumar los lados vecinos \(+\delta Q/T + (-\delta Q/T) = 0\).
🟡 Lina: Exacto. Y los lados de las adiabáticas, ¿qué pasa? — recordemos que "adiabático" significa "sin intercambio de calor" (lo confirmamos en los pasos 2 y 4). Así que sobre una adiabática \(\delta Q = 0\), y \(\delta Q/T = 0\), no contribuye. Al final, todos los lados interiores se eliminan: o bien "los lados sobre isotermas se cancelan con los vecinos" o "los lados sobre adiabáticas son cero de por sí". Lo que queda sin cancelar son solo los lados del perímetro exterior.
🔵 Kai: ¡Ah, con la analogía de los azulejos lo entendí! El interior se cancela todo y solo queda el perímetro exterior. ...Pero ¿eso se cumple sin importar cuán irregular sea la forma del perímetro?
🟡 Lina: Exacto. Como para cada ciclo infinitesimal se cumple \(\delta Q_H/T_H + \delta Q_C/T_C = 0\), al sumar el total solo queda la integral a lo largo del perímetro:
Esto significa que "\(\delta Q_{\text{rev}}/T\) se comporta como una diferencial exacta".
🔵 Kai: ¿Qué es una diferencial exacta?
🟡 Lina: Te lo explico con una analogía de montañismo. La diferencia de altitud solo depende del punto de partida y el de llegada — sin importar qué sendero elijas, \(h(B) - h(A)\) es el mismo valor, ¿verdad? A este tipo de "cambio infinitesimal que no depende del camino" se le llama diferencial exacta. Matemáticamente, es "una cantidad que puede escribirse como el cambio infinitesimal \(dS\) de alguna función \(S\)". En cambio, la distancia recorrida varía según el camino — esa es una diferencial inexacta. \(\delta Q\) es exactamente esto: incluso con el mismo punto inicial y final, su valor cambia según el proceso intermedio.
⚪ Mei: Es decir, \(\delta Q\) en sí es como "la distancia recorrida" y depende del camino, pero al dividir por \(T\) se convierte en algo como "la diferencia de altitud" que no depende del camino.
🟡 Lina: Buena analogía. Que se cumpla \(\oint \delta Q_{\text{rev}}/T = 0\) significa que el resultado de sumar \(\delta Q_{\text{rev}}/T\) de \(A\) a \(B\) no depende del camino. Te doy la explicación intuitiva. Si hay dos caminos de \(A\) a \(B\), el camino I y el camino II, y si los resultados fueran diferentes — yendo por el camino I y volviendo por el inverso del camino II, el valor de una vuelta completa no sería cero. Pero eso contradice \(\oint = 0\). Así que por cualquier camino se obtiene el mismo valor. Lo probaré rigurosamente en la siguiente subsección. Es decir, existe una función \(S\) tal que \(dS = \delta Q_{\text{rev}}/T\) — \(\delta Q_{\text{rev}}/T\) es una diferencial exacta.
Recuerda — \(\delta Q\) en sí era una diferencial inexacta que depende del camino. Pero al dividir por \(T\), se transforma en una cantidad independiente del camino. En matemáticas, a una "cantidad que al multiplicar (o dividir) convierte una diferencial inexacta en exacta" se le llama factor integrante. Aquí \(1/T\) desempeña el papel de factor integrante.
✅ Verificación de comprensión: \(\delta Q\) es una cantidad que depende del camino. ¿Qué propiedad tiene \(\delta Q_{\text{rev}}/T\)?
Respuesta
La integral de \(\delta Q_{\text{rev}}/T\) a lo largo de un proceso reversible no depende del camino (se comporta como una diferencial exacta). Esto permite definir la entropía \(S\) como variable de estado.
La entropía como variable de estado¶
🟡 Lina: Que la integral cíclica de un ciclo reversible sea cero significa que el valor de \(\int \delta Q_{\text{rev}}/T\) a lo largo de un proceso reversible no depende del camino.
Demostrémoslo. Supongamos dos caminos reversibles diferentes I y II del estado \(A\) al estado \(B\). Consideremos el ciclo "camino I de \(A \to B\), inverso del camino II de \(B \to A\)":
Por tanto:
🔵 Kai: ¡Oh, sin importar qué camino se tome se obtiene el mismo valor! Por eso se puede decir que es una "variable de estado".
🟡 Lina: Como no depende del camino, esto define una variable de estado. Fijando un estado de referencia \(O\):
En forma de cambios infinitesimales:
Esta es la definición termodinámica de la entropía \(S\). Aunque \(\delta Q\) era una diferencial inexacta que depende del camino, al dividir por \(T\) se transforma en una variable de estado independiente del camino. \(T\) desempeña el papel de factor integrante.
Relación fundamental de la termodinámica¶
🟡 Lina: Sustituyendo \(\delta Q_{\text{rev}} = T\,dS\) en la primera ley:
Esta es la relación fundamental de la termodinámica. \(dU\), \(dS\) y \(dV\) son todas diferenciales totales de variables de estado; no contiene diferenciales inexactas.
🔵 Kai: Queda elegante. Pero, ¿esta ecuación solo se puede usar en procesos reversibles?
🟡 Lina: Buena pregunta. Como \(U\), \(S\) y \(V\) son todas variables de estado, esta ecuación se cumple como relación entre estados de equilibrio arbitrarios. Sin importar si el proceso es reversible o irreversible, se puede usar universalmente como ecuación que representa la diferencia entre dos estados de equilibrio.
Verificación en el ciclo de Carnot¶
🟡 Lina: Verifiquemos el cambio de entropía en el ciclo de Carnot.
- Expansión isotérmica (\(T_H\)): \(\Delta S_1 = Q_H / T_H\)
- Expansión adiabática: \(\Delta S_2 = 0\) (porque \(\delta Q = 0\))
- Compresión isotérmica (\(T_C\)): \(\Delta S_3 = -Q_C / T_C\)
- Compresión adiabática: \(\Delta S_4 = 0\)
Cambio en una vuelta completa:
⚪ Mei: Efectivamente es cero. Como la entropía es variable de estado, al dar una vuelta vuelve al original.
🔵 Kai: Pero, ¿qué pasa si el proceso es irreversible? ¿No da cero al dar una vuelta?
🟡 Lina: Buena pregunta. En procesos irreversibles \(\delta Q/T < dS\), es decir:
La igualdad se da en procesos reversibles. Para un sistema adiabático (\(\delta Q = 0\)):
Esta es la ley del aumento de la entropía — la formulación de la segunda ley mediante la entropía.
✅ Verificación de comprensión: Escribe la definición termodinámica de la entropía en forma de cambios infinitesimales.
Respuesta
\(dS = \delta Q_{\text{rev}} / T\) (la cantidad infinitesimal de calor añadida al sistema en un proceso reversible dividida por la temperatura).
3.5 La entropía de Boltzmann — De lo micro a lo macro¶
🟡 Lina: Toda la termodinámica hasta aquí no usó ninguna información del mundo microscópico. Definimos "calor", "temperatura" y "entropía" como cantidades puramente macroscópicas. La idea de Boltzmann fue revolucionaria: los fenómenos térmicos macroscópicos se pueden explicar a partir del comportamiento estadístico de las partículas microscópicas.
Microestados y macroestados¶
🔵 Kai: ¿Estadístico?
🟡 Lina: Por ejemplo, piensa en el aire de una habitación. Hay aproximadamente \(10^{23}\) moléculas de aire. Seguir el movimiento de cada una es imposible. Pero "cómo se comporta el conjunto" se puede predecir con probabilidad y estadística.
Aquí distingamos dos conceptos.
- Macroestado: Estado especificado por cantidades medibles macroscópicamente como temperatura, presión, volumen
- Microestado: Estado completamente especificado dando las posiciones y velocidades de todas las partículas (o los estados cuánticos en mecánica cuántica)
Al mismo macroestado generalmente le corresponde un número enorme de microestados.
🔵 Kai: Es decir, ¿aunque no sepamos cómo se mueve una sola molécula, si miramos \(10^{23}\) juntas podemos predecir su comportamiento?
🟡 Lina: Exacto. No puedes predecir el resultado de un dado en una tirada, pero si lo tiras un millón de veces puedes predecir que cada cara saldrá aproximadamente igual, ¿verdad? El mismo principio.
La entropía de Boltzmann¶
🟡 Lina: Boltzmann definió la entropía así:
Donde: - \(\Omega\) = número de microestados correspondientes a un macroestado dado - \(k_B \approx 1.381 \times 10^{-23}\;\text{J/K}\) = constante de Boltzmann. Como se ve por sus unidades J/K (energía ÷ temperatura), desempeña el papel de factor de conversión que conecta el mundo microscópico (energía) con el macroscópico (temperatura) - \(\ln\) = logaritmo natural
🔵 Kai: ¿Por qué se toma el logaritmo?
🟡 Lina: Para la aditividad. Cuando se colocan dos sistemas independientes juntos, el número de microestados del sistema compuesto es el producto:
¿Por qué es un producto? Porque para cada microestado del sistema 1, todos los microestados del sistema 2 son posibles. Es lo mismo que las combinaciones de dos dados: \(6 \times 6 = 36\) resultados.
Al tomar el logaritmo, el producto se convierte en suma:
La entropía se convierte en una cantidad aditiva. La misma propiedad que la energía o el volumen.
⚪ Mei: Tomar el logaritmo es para "convertir multiplicación en suma". No es un truco matemático, sino que viene de una exigencia física.
Entenderlo con el ejemplo de las monedas¶
🟡 Lina: Pensemos con un ejemplo concreto. Lanzamos 4 monedas.
Tabla 3.2: Macroestados y entropía de 4 monedas
| Macroestado (nº de caras) | Nº de microestados \(\Omega\) | Entropía \(S/k_B = \ln\Omega\) |
|---|---|---|
| 0 (todas cruces) | 1 | 0 |
| 1 | 4 | 1.39 |
| 2 | 6 | 1.79 |
| 3 | 4 | 1.39 |
| 4 (todas caras) | 1 | 0 |
🔵 Kai: Todas caras o todas cruces solo tiene 1 forma, pero mitad y mitad tiene 6 formas.
⚪ Mei: Es decir, "2 caras y 2 cruces" es el que tiene mayor \(\Omega\) y donde la entropía es máxima.
🟡 Lina: Así es. En un gráfico se ve de inmediato (Fig. 3.4「Número de microestados y entropía de las monedas」). ¿Ves que tiene un pico en el centro?
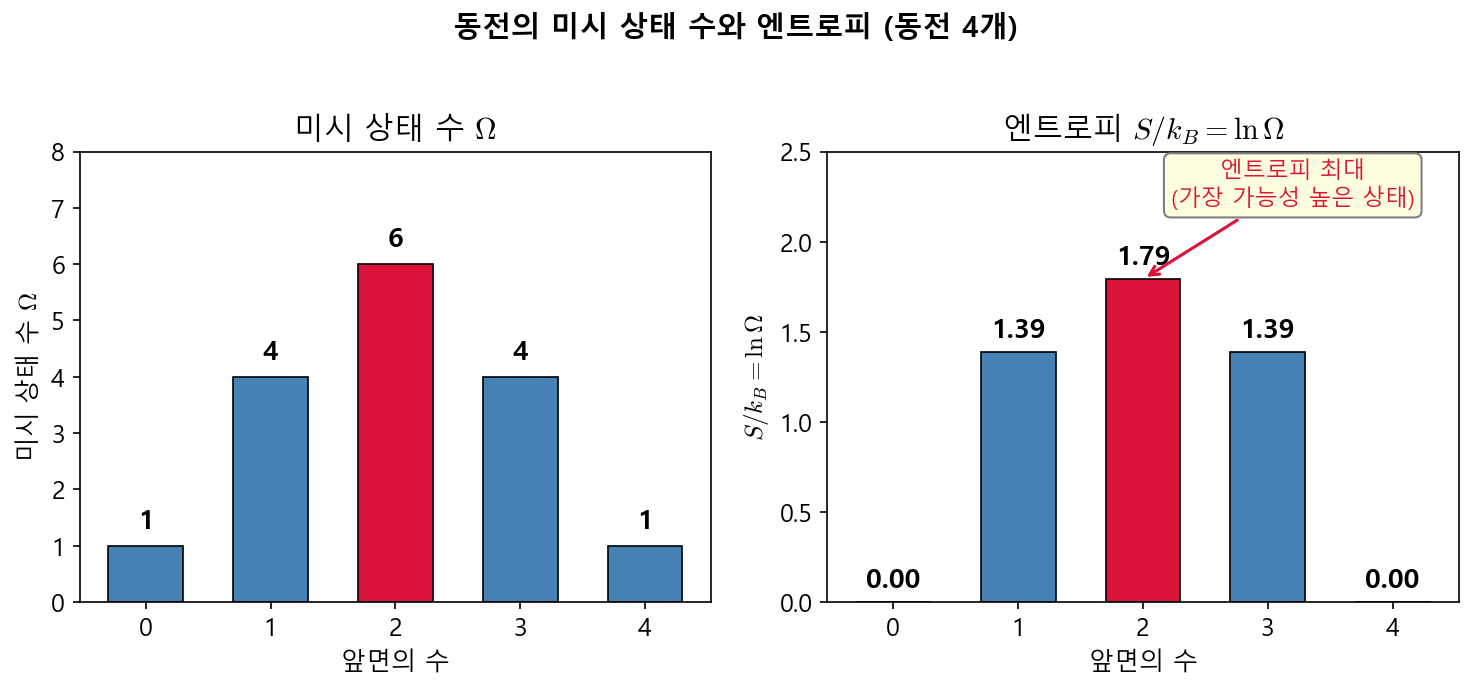
Fig. 3.4: Número de microestados y entropía de las monedas. Para el caso de 4 monedas, se muestra el número de microestados \(\Omega\) y la entropía \(S/k_B = \ln\Omega\) para cada macroestado (número de caras). La entropía es máxima para la distribución más uniforme (2 caras).
✅ Verificación de comprensión: En el ejemplo de 4 monedas, ¿cuál es el macroestado con entropía máxima? ¿Por qué?
Respuesta
El macroestado con 2 caras y 2 cruces. Porque el número de microestados correspondientes \(\Omega = 6\) es máximo (y por tanto \(S = k_B \ln \Omega\) es máximo).
🟡 Lina: Así es. Y cuando son \(10^{23}\) partículas, el \(\Omega\) del "estado distribuido uniformemente" es abrumadoramente mayor que el del "estado concentrado en un lugar". Por eso el sistema se dirige naturalmente hacia el estado con mayor \(\Omega\) — es decir, mayor entropía. En Fig. 3.5「Expansión libre y aumento de la entropía」 puedes ver una imagen concreta. Cuando se retira la pared divisoria, el gas se expande espontáneamente por todo el recipiente. Si nos fijamos solo en "en cuál mitad está cada partícula, izquierda o derecha", el estado inicial (todas las partículas en la mitad izquierda) tiene solo 1 configuración, pero en el estado de equilibrio, como cada partícula puede estar en cualquiera de los dos lados, el número de configuraciones explota a \(2^N\).
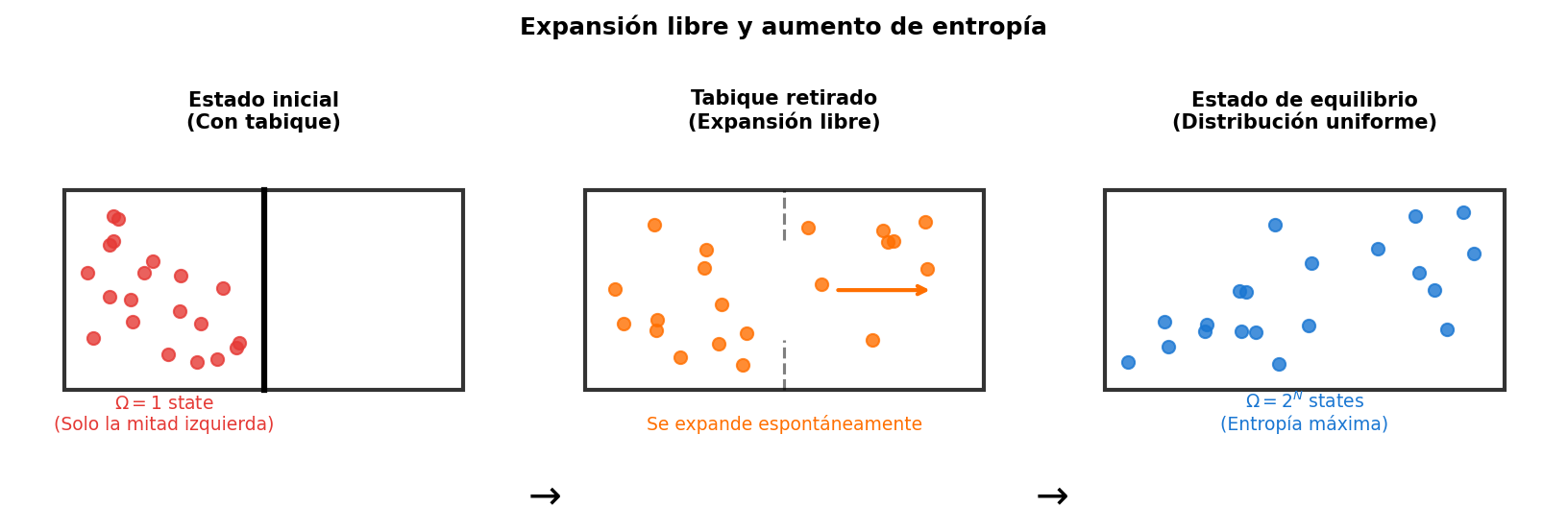
Fig. 3.5: Expansión libre y aumento de la entropía. Al retirar la pared divisoria, el gas se expande espontáneamente por todo el volumen. Fijándose solo en "en cuál mitad está cada partícula", el estado inicial (todas las partículas en la mitad izquierda) tiene 1 sola configuración, pero en el estado de equilibrio (distribución uniforme), como cada partícula puede estar en cualquiera de los dos lados, el número de configuraciones aumenta a \(2^N\).
Por qué se cumple la segunda ley¶
🔵 Kai: ¿Esa es la razón de que "el calor fluye de caliente a frío"?
🟡 Lina: Sí. El calor fluye de caliente a frío porque así aumenta el número total de microestados \(\Omega\) del conjunto. No está prohibido, simplemente la probabilidad de que ocurra en dirección contraria es astronómicamente pequeña.
🔵 Kai: Es decir, no es una "prohibición" sino "casi con certeza ocurre así"... Pero "casi con certeza", ¿cuán pequeña es la probabilidad concretamente? ¿Como ganar la lotería? ¿O es de un orden de magnitud mucho menor?
🟡 Lina: Estimémoslo concretamente. Si hay \(N\) moléculas de gas en una caja, la probabilidad de que todas se concentren en la mitad izquierda es:
Si \(N = 10^{23}\):
🔵 Kai: \(10^{-3 \times 10^{22}}\)... Ni de lejos se compara con la lotería. Una probabilidad con \(10^{22}\) ceros... es prácticamente "no ocurre".
🟡 Lina: Durante toda la edad del universo (unos \(10^{10}\) años \(\approx 10^{17}\) segundos), incluso intentándolo una vez por segundo, nunca ocurriría.
🔭 Nota de filosofía de la ciencia: La segunda ley no es una "ley que jamás se rompe" sino una "ley estadística cuya probabilidad de violación es astronómicamente pequeña". Esto tiene un carácter esencialmente diferente de las leyes deterministas como las de Newton. Juzga por ti mismo — ¿se puede afirmar que "un evento con probabilidad \(10^{-10^{22}}\) no ocurre"?
📝 Ejercicios:
- Número de microestados y entropía de las monedas → Problema M-1. Entropía de una moneda
✅ Verificación de comprensión: Escribe la definición de la entropía de Boltzmann e indica qué representa \(\Omega\).
Respuesta
\(S = k_B \ln \Omega\). \(\Omega\) es el número de microestados (el número total de configuraciones microscópicas que dan el mismo estado macroscópico).
✅ Verificación de comprensión: Explica la razón por la que el calor fluye de caliente a frío utilizando el número de microestados.
Respuesta
Porque cuando el calor fluye de caliente a frío, el número total de microestados \(\Omega\) del conjunto aumenta. No está prohibido que ocurra en sentido contrario, simplemente su probabilidad es astronómicamente pequeña.
3.6 Número de microestados del gas ideal — Cálculo concreto¶
🟡 Lina: Como \(S = k_B \ln \Omega\) puede parecer abstracto, calculemos \(\Omega\) concretamente para un gas ideal.
Planteamiento del problema¶
🟡 Lina: Consideremos \(N\) partículas idénticas (masa \(m\)) encerradas en un volumen \(V\), con energía total \(E\).
En mecánica clásica, el estado de cada partícula se especifica completamente con 6 números: posición \((x, y, z)\) y momento \((p_x, p_y, p_z)\). El momento es "masa × velocidad", y es una cantidad vectorial con componentes \(p_x = mv_x\), \(p_y = mv_y\), \(p_z = mv_z\) en cada dirección.
🔵 Kai: ¿No basta solo con la posición?
🟡 Lina: No basta. Aunque dos partículas estén en la misma posición, si una se mueve rápido y la otra está parada, sus estados son diferentes, ¿verdad? Por eso necesitamos tanto "dónde está" como "cómo se mueve". Para 1 partícula son \((x, y, z, p_x, p_y, p_z)\), 6 números — si consideramos un espacio de 6 dimensiones con estos como ejes coordenados, un solo punto en ese espacio representa completamente "dónde está la partícula y cómo se mueve". Para 2 partículas se necesitan \(6 \times 2 = 12\) números para determinar el estado del sistema completo. Para \(N\) partículas se necesitan \(6N\) variables en total. Al espacio de \(6N\) dimensiones con estas \(6N\) variables como ejes coordenados se le llama espacio fásico (phase space). "Fásico" no tiene relación con la fase de una onda — hay varias teorías sobre el origen del nombre, pero aquí piensa que significa "espacio que representa completamente el estado del sistema". Un punto del espacio fásico corresponde a un microestado del sistema completo.
⚪ Mei: Es decir, para \(10^{23}\) partículas, un solo punto en un espacio de \(6 \times 10^{23}\) dimensiones describe completamente el sistema. Son dimensiones descomunales pero el concepto es simple.
La restricción de energía¶
🟡 Lina: En un gas ideal no hay interacción entre partículas, así que la energía total es la suma de energías cinéticas:
Para ver mejor la estructura de esta expresión, renombremos las \(3N\) componentes de momento como \(\xi_1, \xi_2, \ldots, \xi_{3N}\) (\(\xi_1 = p_{x,1}\), \(\xi_2 = p_{y,1}\), \(\xi_3 = p_{z,1}\), \(\xi_4 = p_{x,2}\), …). Entonces la condición de energía total es:
🔵 Kai: \(\xi_1^2 + \xi_2^2 + \cdots = 2mE\) parece como una esfera. En 3 dimensiones \(x^2 + y^2 + z^2 = R^2\) es una esfera...
🟡 Lina: Buena intuición. Exactamente así, es la ecuación de una esfera en \(3N\) dimensiones — de radio \(\sqrt{2mE}\). Es decir, una vez determinada la energía, la "distribución" de los momentos queda restringida a una superficie esférica, y cuanto mayor sea la energía, mayor es la esfera y más estados hay. La restricción de energía se traduce en una restricción geométrica — el tamaño de la esfera.
⚪ Mei: Es decir, la restricción de energía se traduce en la condición geométrica del "radio de la esfera".
🟡 Lina: Así es. He dibujado una imagen en Fig. 3.6「Esfera en el espacio de momentos y número de microestados」.
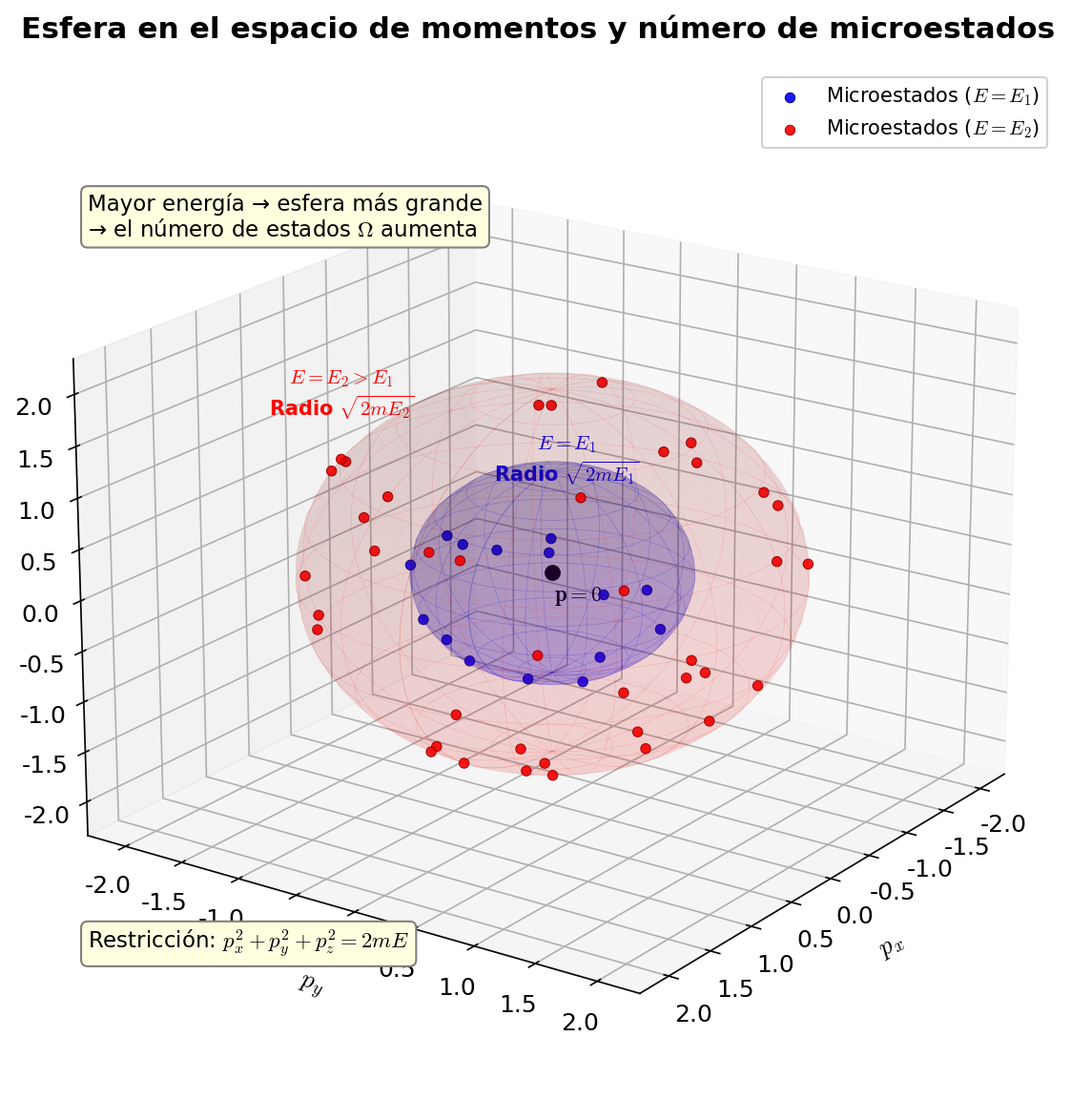
Fig. 3.6: Esfera en el espacio de momentos y número de microestados. Los microestados de un gas ideal corresponden a la esfera (su interior) de radio \(\sqrt{2mE}\) en el espacio de momentos. Al aumentar la energía, la esfera crece y aumenta el número de microestados.
Los estados con energía exactamente \(E\) están sobre la superficie esférica, pero al contar el número de microestados usamos el volumen interior de la esfera (todos los estados con energía \(\leq E\)). En realidad, en alta dimensión casi todo el volumen de la esfera se concentra cerca de la superficie. Intuitivamente, el volumen de una esfera \(d\)-dimensional es proporcional a \(R^d\), así que la fracción del volumen de una esfera de radio \(0.99R\) respecto a una de radio \(R\) es \((0.99)^d\) — para \(d = 3\) es aproximadamente 97%, pero para \(d = 10^{23}\) es \((0.99)^{10^{23}} \approx 0\), es decir, prácticamente todo el volumen se concentra en la capa exterior del 1%. Por eso contar con el volumen interior o con el área superficial da el mismo resultado. Aquí procederemos asumiendo que es proporcional al volumen interior de la esfera.
🔵 Kai: Que en alta dimensión el volumen se concentre en la superficie... es contraintuitivo. Pero cuando me dicen \((0.99)^{10^{23}} \approx 0\) me convenzo.
Volumen de una esfera en \(3N\) dimensiones¶
🔵 Kai: ¿Cómo se calcula el volumen de una esfera de \(3N\) dimensiones? En 3 dimensiones es \(\frac{4}{3}\pi R^3\) pero...
🟡 Lina: Buena pregunta. Para obtener el número de microestados necesitamos calcular el volumen de la esfera de radio \(\sqrt{2mE}\) en el espacio de momentos. En 3 dimensiones el volumen de la esfera es \(\frac{4}{3}\pi R^3\) — proporcional a \(R^3\). Extendiendo la misma idea a \(d\) dimensiones, se puede calcular mediante integrales múltiples (omito la derivación) y se obtiene:
Aquí \(\Gamma\) es la función gamma — una función que extiende el factorial \(n!\) a valores no enteros, y para enteros \(n\) satisface \(\Gamma(n+1) = n!\). ¿Por qué aparece \(\pi\)? Recuerda el área del círculo en 2 dimensiones \(\pi R^2\) — \(\pi\) viene de "dar la vuelta completa en la dirección angular". En dimensiones superiores es igual: al calcular el volumen de la esfera mediante integrales múltiples, la parte que integra en todas las direcciones angulares produce potencias de \(\pi\), y la integral en la dirección radial (distancia al centro) produce naturalmente el factorial (o su generalización, la función gamma).
⚪ Mei: \(\pi\) viene de la geometría de "dar la vuelta completa", y \(\Gamma\) de "acumular en la dirección radial" — cada uno con un origen diferente.
🟡 Lina: Exacto. No necesitas memorizar la fórmula — lo importante es la parte proporcional a \(R^d\). Es decir, cuanto mayor sea la dimensión \(d\), el volumen crece explosivamente con solo un pequeño aumento del radio \(R\). Verifiquémoslo en bajas dimensiones. Para \(d = 1\): \(V_1 = \frac{\pi^{1/2}}{\Gamma(3/2)} R = \frac{\sqrt{\pi}}{\frac{1}{2}\sqrt{\pi}} R = 2R\) — es un segmento de longitud \(2R\), correcto. Para \(d = 2\): \(V_2 = \frac{\pi}{\Gamma(2)} R^2 = \pi R^2\) — el área del círculo. Para \(d = 3\): \(V_3 = \frac{\pi^{3/2}}{\Gamma(5/2)} R^3 = \frac{\pi^{3/2}}{\frac{3}{4}\sqrt{\pi}} R^3 = \frac{4}{3}\pi R^3\) — el volumen de la esfera. Todo cuadra.
🔵 Kai: \(\Gamma(d/2 + 1)\), si \(d\) es impar entran valores como \(\Gamma(3/2)\) o \(\Gamma(5/2)\) que no son enteros, ¿se puede calcular eso?
🟡 Lina: Buena observación. La función gamma es una función que extiende el factorial \(n!\) a valores no enteros, y satisface la relación de recurrencia \(\Gamma(n+1) = n \cdot \Gamma(n)\). Se conoce el valor \(\Gamma(1/2) = \sqrt{\pi}\), y a partir de ahí se calcula \(\Gamma(3/2) = \frac{1}{2}\sqrt{\pi}\), \(\Gamma(5/2) = \frac{3}{4}\sqrt{\pi}\) sucesivamente. Pero lo importante ahora no son los detalles de estos coeficientes, sino que el volumen es proporcional a \(R^d\) — es decir, cuanto mayor sea la dimensión \(d\), el volumen crece explosivamente con un pequeño aumento del radio. Las propiedades detalladas de la función gamma las aprenderemos cuando sean necesarias. Sustituyendo \(R = \sqrt{2mE}\):
Construcción del número de microestados¶
🟡 Lina: Ahora consideremos los grados de libertad de posición. Una partícula puede estar en cualquier lugar del volumen \(V\), así que las "opciones" de posición son proporcionales a \(V\). Para 2 partículas, la partícula 1 puede estar en cualquier lugar de \(V\), la partícula 2 también independientemente en cualquier lugar de \(V\) — así que \(V \times V = V^2\). Para \(N\) partículas: \(V^N\). Es decir, los grados de libertad de posición contribuyen con un factor \(V^N\).
🔵 Kai: ¿Si multiplicamos el volumen de la esfera en el espacio de momentos por \(V^N\) obtenemos el número de microestados?
🟡 Lina: Hacen falta un par de correcciones más. Primero, el "volumen" del espacio fásico es continuo, así que tal cual no da un "número" de estados — el volumen es un valor real, no se puede decir "cuántas configuraciones hay".
🔵 Kai: Cierto. Si te dicen "el área es 10 metros cuadrados", no puedes decir "cuántas celdas" sin fijar el tamaño de la celda.
🟡 Lina: Exacto. Para contar estados hay que dividir el espacio fásico en alguna unidad mínima y contar "cuántas celdas". Es como decidir el tamaño de las celdas de un papel cuadriculado. Entonces, ¿qué determina el tamaño de la celda? Aquí entra la mecánica cuántica (que estudiaremos en detalle en los próximos capítulos). La mecánica cuántica tiene un principio que dice "no se pueden determinar simultáneamente con exactitud completa la posición y el momento" — por ejemplo, si intentas medir con precisión la posición de una partícula, la información del momento se difumina. Como consecuencia de este principio, en el espacio fásico existe un área mínima por debajo de la cual no se puede distinguir, una "celda mínima". La constante que determina esa área mínima es \(h\) (\(h \approx 6.626 \times 10^{-34}\;\text{J·s}\), llamada constante de Planck) — una constante fundamental con la que la naturaleza dice "no distinguiré más fino que esto". Por qué tiene este valor y por qué existe este principio lo estudiaremos en detalle en los próximos capítulos.
🔵 Kai: Es decir, el valor concreto de \(h\) está determinado experimentalmente, y por ahora basta aceptarlo como "el tamaño de celda que la naturaleza ha fijado", ¿verdad?
🟡 Lina: Exacto. En cada dirección, el valor mínimo de \(\Delta x \cdot \Delta p_x\) está determinado en aproximadamente \(h\). Una partícula tiene posición y momento en cada una de las 3 direcciones \(x, y, z\), así que la celda mínima es \(h \times h \times h = h^3\) (es un producto porque cada dirección es independiente). Para \(N\) partículas: \(h^{3N}\). Por eso dividimos el volumen del espacio fásico por \(h^{3N}\) para convertirlo en "número de estados".
Además, las partículas del mismo tipo son indistinguibles al intercambiarlas — esto es un requisito que viene de la mecánica cuántica, que estudiaremos en detalle en los próximos capítulos. Intuitivamente, no se puede "poner etiquetas" al átomo de helio A y al átomo de helio B — como ambos tienen exactamente las mismas propiedades, intercambiarlos no produce ninguna diferencia física detectable. Por ahora acepta que "no se pueden etiquetar y distinguir partículas del mismo tipo". Intercambiar la partícula 1 y la 2 da el mismo microestado. Hay \(N!\) formas de permutar \(N\) partículas, así que para eliminar la duplicación dividimos por \(N!\). Combinando todo:
(Cuando \(N\) es grande, \(\Gamma(3N/2+1)\) se puede tratar con la aproximación de Stirling que presentaré en la siguiente subsección, así que no hay que preocuparse de si es entero o no.)
🔵 Kai: Es complicado...
🟡 Lina: Lo importante es la dependencia en \(E\) y \(V\). Viendo como función solo de \(E\) y \(V\) con \(N\) fijo (todos los factores que dependen de \(N\) se tratan como constantes):
⚪ Mei: Aumentar el volumen o aumentar la energía — cualquiera de los dos aumenta el número de microestados. Un resultado simple que tranquiliza.
Cálculo de la entropía¶
🟡 Lina: Tomando el logaritmo:
Tal cual, \(\ln(N!)\) y \(\ln\Gamma(3N/2+1)\) son difíciles de manejar. Aquí usamos la aproximación de Stirling. Cuando \(n\) es grande, el logaritmo de \(n!\) es:
Esto se puede aproximar porque \(\ln n! = \ln 1 + \ln 2 + \cdots + \ln n\) se aproxima por la integral \(\int_1^n \ln x\,dx = n\ln n - n + 1 \approx n\ln n - n\).
🔵 Kai: Que el logaritmo del factorial se pueda aproximar por \(n \ln n - n\) es muy útil. Calcular directamente \(10^{23}!\) sería imposible.
🟡 Lina: Aplicándola: \(\ln(N!) \approx N\ln N - N\), \(\ln\Gamma(3N/2+1) \approx \frac{3N}{2}\ln\frac{3N}{2} - \frac{3N}{2}\). Reorganizando: \(N\ln V - N\ln N = N\ln(V/N)\) y \(\frac{3N}{2}\ln(2mE) - \frac{3N}{2}\ln\frac{3N}{2} = \frac{3N}{2}\ln\frac{4mE}{3N}\). Además, \(\ln\frac{4mE}{3N} = \ln\frac{E}{N} + \ln\frac{4m}{3}\), donde la parte \(\ln\frac{4m}{3}\) es una constante que no depende ni de \(E\) ni de \(V\), así que se puede agrupar con los demás términos constantes que incluyen \(m\) y \(h\) (nótese que el argumento del logaritmo se vuelve adimensional al combinar con estas constantes). Finalmente se obtiene el resultado conocido como la fórmula de Sackur-Tetrode:
Donde (constante) incluye todos los términos construidos a partir de \(m\), \(h\), \(k_B\), etc. Podrías pensar "el argumento de \(\ln(E/N)\) tiene dimensiones de energía, ¿está bien meterlo en un logaritmo?", pero en realidad dentro de la constante hay términos como \(\frac{3}{2}\ln(4\pi m / (3h^2))\) y al combinarlos el argumento del \(\ln\) se vuelve adimensional. Sin embargo, al derivar parcialmente respecto a \(E\) o \(V\), estos términos constantes desaparecen y no afectan los cálculos siguientes. Así que por ahora piensa "no hay que preocuparse por la parte constante".
⚪ Mei: Cuanto mayor es \(E\), cuanto mayor es \(V\), mayor es la entropía. Coincide con la intuición.
🟡 Lina: Así es. Si la energía aumenta, la esfera en el espacio de momentos crece; si el volumen aumenta, el espacio de posiciones se amplía. Ambos aumentan el número de microestados.
✅ Verificación de comprensión: ¿Cómo depende el número de microestados \(\Omega\) del gas ideal de la energía \(E\)?
Respuesta
\(\Omega \propto E^{3N/2}\). Porque al aumentar la energía, el volumen de la esfera en el espacio de momentos aumenta.
3.7 Significado estadístico de la temperatura¶
🟡 Lina: Usando la entropía de Boltzmann, también se puede dar un significado estadístico a la temperatura.
Derivación a partir de la condición de equilibrio térmico de dos sistemas¶
🟡 Lina: Consideremos una situación donde dos sistemas (sistema 1 y sistema 2) pueden intercambiar energía. La energía total se conserva:
El número de microestados del sistema compuesto es:
Aquí introduzco una hipótesis importante. El principio de igual probabilidad a priori: en un sistema aislado, todos los microestados que satisfacen las condiciones como la energía se realizan con igual probabilidad.
🔵 Kai: ¿Por qué se puede decir que son equiprobables?
🟡 Lina: Es una pregunta profunda. No hay una demostración rigurosa. Pero el argumento de simetría de que "no hay razón para favorecer un microestado particular", junto con el hecho de que los resultados derivados de esta hipótesis coinciden con los experimentos, sustentan este principio. Piénsalo como la hipótesis de partida de la mecánica estadística.
🔵 Kai: ¿Y eso cómo lleva a la maximización de \(\Omega\)?
🟡 Lina: Si cada microestado es equiprobable, la probabilidad de que se realice un cierto macroestado es proporcional al número de microestados \(\Omega\) correspondientes a ese macroestado. Por tanto, el macroestado con \(\Omega\) máximo se realiza con la mayor probabilidad — es decir, el sistema se dirige hacia la distribución de energía que maximiza \(\Omega_{\text{total}}\).
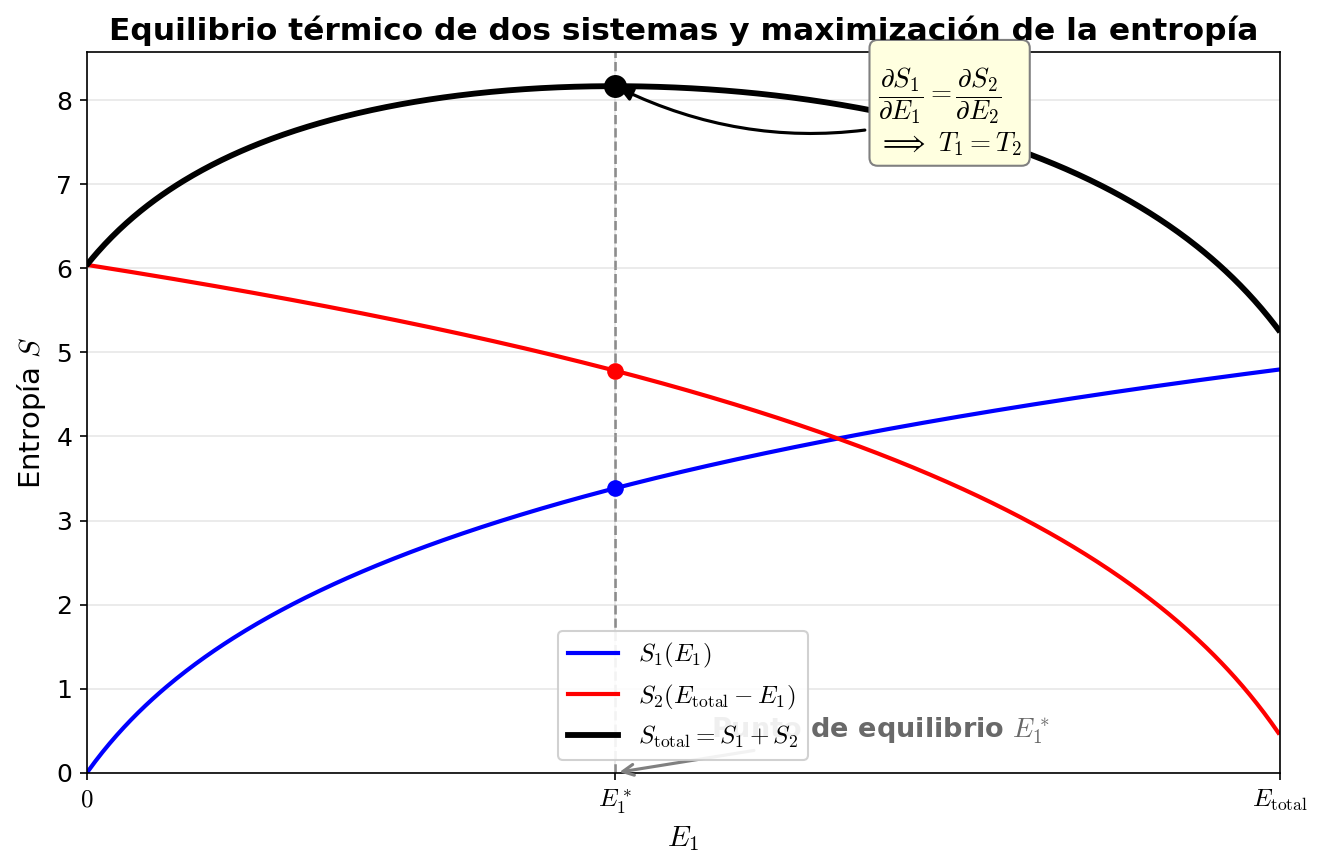
Fig. 3.7: Equilibrio térmico y maximización de la entropía. Cuando dos sistemas intercambian energía, se alcanza el equilibrio en el punto donde la entropía total \(S_{\text{total}} = S_1(E_1) + S_2(E_{\text{total}} - E_1)\) es máxima. En ese punto \(\partial S_1/\partial E_1 = \partial S_2/\partial E_2\), es decir \(T_1 = T_2\).
🟡 Lina: Solo necesitamos encontrar la condición que maximiza \(\Omega_{\text{total}}\). He dibujado una imagen en Fig. 3.7「Equilibrio térmico y maximización de la entropía」. Como es más fácil calcular con logaritmos, escribámoslo en términos de entropía:
Derivamos respecto a \(E_1\) e igualamos a cero:
Como \(E_2 = E_{\text{total}} - E_1\), tenemos \(\partial E_2 / \partial E_1 = -1\). Por tanto:
🔵 Kai: ¡En equilibrio, \(\partial S / \partial E\) es igual en ambos sistemas!
🟡 Lina: Así es. Y sabemos que "en equilibrio térmico las temperaturas son iguales". Por tanto, \(\partial S / \partial E\) debe estar relacionado con la temperatura. En efecto:
Esta es la definición estadística de la temperatura.
✅ Verificación de comprensión: Expresa la condición de equilibrio térmico cuando dos sistemas intercambian energía, utilizando la entropía.
Respuesta
\(\frac{\partial S_1}{\partial E_1} = \frac{\partial S_2}{\partial E_2}\) (la derivada de la entropía respecto a la energía es igual en ambos sistemas). Esto corresponde a la igualdad de temperaturas.
Verificación con el gas ideal¶
🟡 Lina: La entropía del gas ideal que obtuvimos antes era \(S = k_B N \left[\frac{3}{2}\ln \frac{E}{N} + \ln\frac{V}{N} + (\text{constante})\right]\). Al derivar parcialmente respecto a \(E\), como \(\ln(E/N) = \ln E - \ln N\), la parte \(\ln N\) desaparece como constante. Derivemos:
Reorganizando:
🔵 Kai: \(E = \frac{3}{2}Nk_BT\)... ¡es la misma ecuación que usamos en el cálculo del proceso adiabático! ¿Lo que entonces usamos sin derivar, aquí lo hemos deducido?
🟡 Lina: Exacto. Antes lo usamos como "hecho conocido", pero ahora acabamos de derivarlo desde la mecánica estadística. El \(3\) es el número de grados de libertad de cada partícula (movimiento en las direcciones \(x, y, z\)), y cada grado de libertad tiene una energía de \(\frac{1}{2}k_B T\). Esto se llama el teorema de equipartición de la energía.
🔵 Kai: Entonces, si una molécula también puede rotar, ¿aumentan los grados de libertad y a la misma temperatura la energía es mayor?
🟡 Lina: Exacto. Una molécula diatómica suma 2 grados de libertad de rotación, dando \(E = \frac{5}{2}Nk_BT\). El poder del teorema de equipartición. Además, partiendo solo de la definición estadística \(1/T = \partial S/\partial E\), todas estas fórmulas conocidas se pueden derivar. De manera similar, de la derivada de la entropía respecto a \(V\) se obtiene la presión:
Reorganizando:
🔵 Kai: ¡La ecuación de estado del gas ideal! Pero espera. Al calcular \(\partial S/\partial V\), fijamos \(E\). ¿Por qué se fija la energía y no la temperatura?
🟡 Lina: Buena pregunta. Aquí consideramos un sistema aislado (sin intercambio de energía con el exterior), así que \(E\) es constante. La temperatura es una cantidad derivada de \(E\), por lo que es natural fijar \(E\) como punto de partida.
⚪ Mei: Es decir, obtenemos la entropía a partir de \(S = k_B \ln \Omega\), derivamos parcialmente respecto a \(E\) y sale la temperatura, derivamos respecto a \(V\) y sale la presión.
🟡 Lina: Exacto. Contando microestados para obtener la entropía, y simplemente derivando parcialmente, se pueden deducir la temperatura, la presión y la ecuación de estado. Todas las cantidades macroscópicas emergen del conteo de microestados — ese es el poder de la mecánica estadística.
✅ Verificación de comprensión: Al derivar \(E = \frac{3}{2}Nk_BT\) a partir de la entropía del gas ideal, ¿qué operación se realiza?
Respuesta
Se calcula la derivada parcial de la entropía \(S = k_B N [\frac{3}{2}\ln E + \cdots]\) respecto a la energía \(E\) para obtener \(1/T = \partial S/\partial E\), y se despeja \(E\).
Significado intuitivo de la temperatura¶
🟡 Lina: Expresando en palabras la definición de temperatura \(1/T = \partial S / \partial E\):
Cuanto menor es la temperatura de un sistema, más aumenta la entropía (logaritmo del número de estados) al añadirle un poco de energía.
Un objeto a alta temperatura ya es "desordenado", así que al añadir energía no se abren tantos nuevos estados. Un objeto a baja temperatura aún es "ordenado", así que con poca energía se abren muchos nuevos estados.
🔵 Kai: Cuanto más baja la temperatura, mayor es el "margen de mejora". El efecto de transferir energía es mayor.
🟡 Lina: Así es. Confirmémoslo visualmente con el gráfico de Fig. 3.8「Significado estadístico de la temperatura」. En el gráfico \(S\)-\(E\), para un mismo sistema, en la región de baja energía (baja temperatura) la pendiente \(\partial S/\partial E\) es empinada, y en la región de alta energía (alta temperatura) la pendiente es suave.
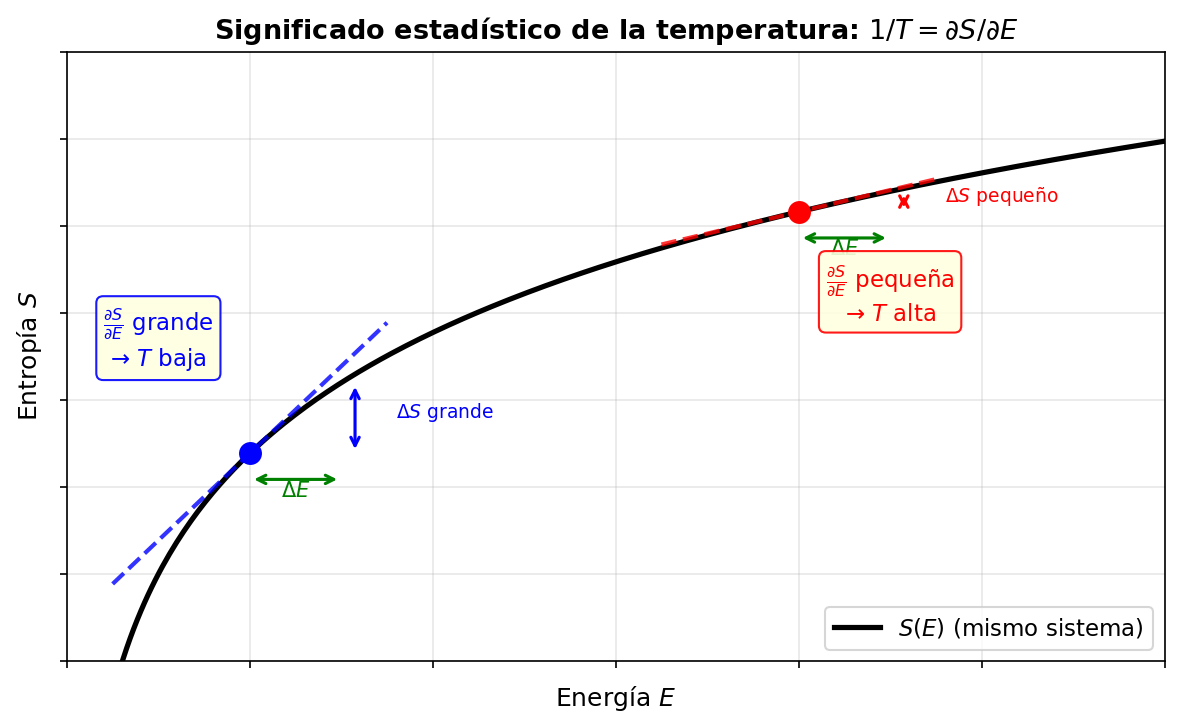
Fig. 3.8: Significado estadístico de la temperatura. La pendiente del gráfico \(S\)-\(E\), \(\partial S/\partial E = 1/T\), da el inverso de la temperatura. La región de pendiente empinada (azul) corresponde a baja temperatura: el mismo \(\Delta E\) produce un gran aumento de entropía. La región de pendiente suave (roja) corresponde a alta temperatura: al añadir energía la entropía apenas aumenta.
Por eso, cuando la energía fluye del sistema de alta temperatura al de baja temperatura, la disminución de entropía del lado caliente es menor que el aumento de entropía del lado frío, y en total la entropía aumenta.
🔵 Kai: ¡Por eso el calor fluye de caliente a frío! Pero, si el sistema fuera pequeño con solo unas 10 partículas, ¿se podría observar el flujo en dirección contraria?
🟡 Lina: Perspicaz. De hecho, en sistemas a escala nanométrica se observan desviaciones de la segunda ley como "fluctuaciones". Cuando el número de partículas es pequeño, la razón de \(\Omega\) ya no es abrumadora. Pero en sistemas macroscópicos (\(N \sim 10^{23}\)) prácticamente no ocurre.
🔵 Kai: Ah, entonces la segunda ley es una "ley aproximada" que se cumple precisamente porque hay muchas partículas.
🟡 Lina: Exacto. Las leyes de Newton "se cumplen determinísticamente para cada partícula individual", pero la segunda ley es "una ley estadística que solo se cumple de facto cuando \(N\) es grande" — el carácter de la ley es fundamentalmente diferente. Las leyes físicas tienen al menos dos tipos de "manera de cumplirse": leyes deterministas que se cumplen rigurosamente incluso para una sola partícula, y leyes estadísticas que solo se cumplen de facto cuando \(N\) es grande. La segunda ley es de este último tipo: a escala nanométrica se ven violaciones, pero a escala macroscópica parece absoluta.
⚪ Mei: Es decir, incluso bajo el nombre de "ley física", el mecanismo por el que se cumplen es totalmente diferente. Leyes deterministas y leyes estadísticas — es necesario distinguirlas.
📝 Ejercicios:
- Derivación de la igualdad de temperaturas a partir de la condición de equilibrio térmico → Problema M-2. Definición estadístico-mecánica de la temperatura
✅ Verificación de comprensión: Escribe la definición estadística de la temperatura.
Respuesta
\(\frac{1}{T} = \frac{\partial S}{\partial E}\bigg|_{V,N}\) (la derivada parcial de la entropía respecto a la energía es el inverso de la temperatura).
✅ Verificación de comprensión: ¿Cómo cambia el número de microestados cuando se añade energía a un objeto a baja temperatura?
Respuesta
Aumenta mucho (como \(\partial S / \partial E\) es grande, \(T\) es pequeña).
3.8 Energía libre \(F = U - TS\)¶
🟡 Lina: Por último, voy a introducir un concepto más importante. La energía libre de Helmholtz.
Motivación: Sistemas a temperatura constante¶
🟡 Lina: En el laboratorio, frecuentemente se mantiene la temperatura constante poniendo el sistema en contacto con un gran baño térmico. En este caso, la entropía del sistema no es constante (porque intercambia energía con el baño). En su lugar, la temperatura \(T\) es constante.
La cantidad que determina "a qué estado se asienta el sistema" en esta situación es la energía libre:
Principio de minimización de \(F\)¶
🟡 Lina: Si consideramos el sistema total (sistema + baño térmico), la entropía total aumenta (segunda ley). Aquí asumimos que el volumen del sistema es constante (no realiza trabajo) — la situación típica de un sistema en un recipiente sumergido en un baño térmico. Bajo esta condición, derivemos, a partir del cambio de entropía total, una condición escrita solo en términos de cantidades del sistema.
🟡 Lina: Calculemos el cambio de entropía del baño térmico. Hay dos puntos clave:
- El baño térmico es muy grande, así que al recibir energía del sistema su temperatura apenas cambia. Imagina verter una gota de agua hirviendo en una piscina enorme — la temperatura de la piscina apenas cambia, ¿verdad? Es decir, el baño siempre está prácticamente en equilibrio, y el intercambio de energía con el sistema es "solo un cambio infinitesimal" para el baño. Por eso, para el baño se puede usar directamente \(dS = \delta Q_{\text{rev}}/T\) — para el calor \(\Delta Q_{\text{bath}}\) recibido por el baño se puede escribir \(\Delta S_{\text{bath}} = \Delta Q_{\text{bath}}/T\). (Estrictamente hablando se necesita la condición de "proceso cuasiestático sin disipación es reversible", pero como para el baño enorme es un cambio infinitesimal, se puede considerar que esta condición se satisface automáticamente.)
- Como el volumen es constante, el sistema no realiza trabajo (\(\Delta W = p\Delta V = 0\)). De la primera ley \(\Delta U = \Delta Q - \Delta W\) con \(\Delta W = 0\): el cambio de energía del sistema es \(\Delta U_{\text{sys}} = \Delta Q_{\text{sys}}\) (el calor recibido por el sistema se convierte directamente en cambio de energía interna). Por conservación de la energía, el calor recibido por el sistema es igual al perdido por el baño: \(\Delta Q_{\text{bath}} = -\Delta Q_{\text{sys}} = -\Delta U_{\text{sys}}\)
🔵 Kai: Entiendo, lo que el sistema gana en energía es lo que pierde el baño — conservación de la energía.
🟡 Lina: Resumiendo:
Exigiendo \(\Delta S_{\text{total}} \geq 0\):
Multiplicando ambos lados por \(-T\) (como \(T > 0\), la desigualdad se invierte):
Como la temperatura \(T\) es constante (el baño fija la temperatura), \(\Delta U - T\Delta S = \Delta(U - TS)\). Es decir:
🟡 Lina: Es decir, a temperatura constante y volumen constante, el sistema cambia en la dirección que minimiza la energía libre \(F\).
⚪ Mei: Al reescribir la ley de aumento de la entropía solo en términos de cantidades del sistema, se convierte en la disminución de \(F\).
Significado físico de \(F\)¶
🟡 Lina: Piensa en el significado de \(F = U - TS\).
- \(U\) quiere bajar la energía (estabilidad)
- \(-TS\) quiere aumentar la entropía (desorden)
\(F\) resume en una sola cantidad la competencia entre estos dos. A baja temperatura (\(T\) pequeña), la minimización de energía domina y el sistema prefiere estados ordenados. A alta temperatura (\(T\) grande), la maximización de entropía domina y el sistema prefiere estados desordenados.
✅ Verificación de comprensión: En la energía libre \(F = U - TS\), ¿cuál de los dos términos domina a baja temperatura y cuál a alta temperatura?
Respuesta
A baja temperatura domina \(U\) (minimización de energía) y el sistema prefiere estados ordenados; a alta temperatura domina \(-TS\) (maximización de entropía) y el sistema prefiere estados desordenados.
🔵 Kai: Que el agua se vuelva hielo (orden) a baja temperatura y vapor (desorden) a alta temperatura es resultado de esta competencia. Pero, justo en el límite — la temperatura a la que el hielo se derrite y demás — ¿cómo se determina?
🟡 Lina: Buena pregunta. Es la temperatura a la que el estado que minimiza \(F\) cambia de "estado ordenado" a "estado desordenado", y esa es la temperatura de transición de fase. Al subir la temperatura, en cierto punto el término \(-TS\) empieza a ganar al término \(U\), y el estado desordenado pasa a tener menor \(F\) — ese instante de cambio es la transición de fase. En Fig. 3.9「Competencia entre energía y entropía en la energía libre」 muestro una imagen de esta competencia.
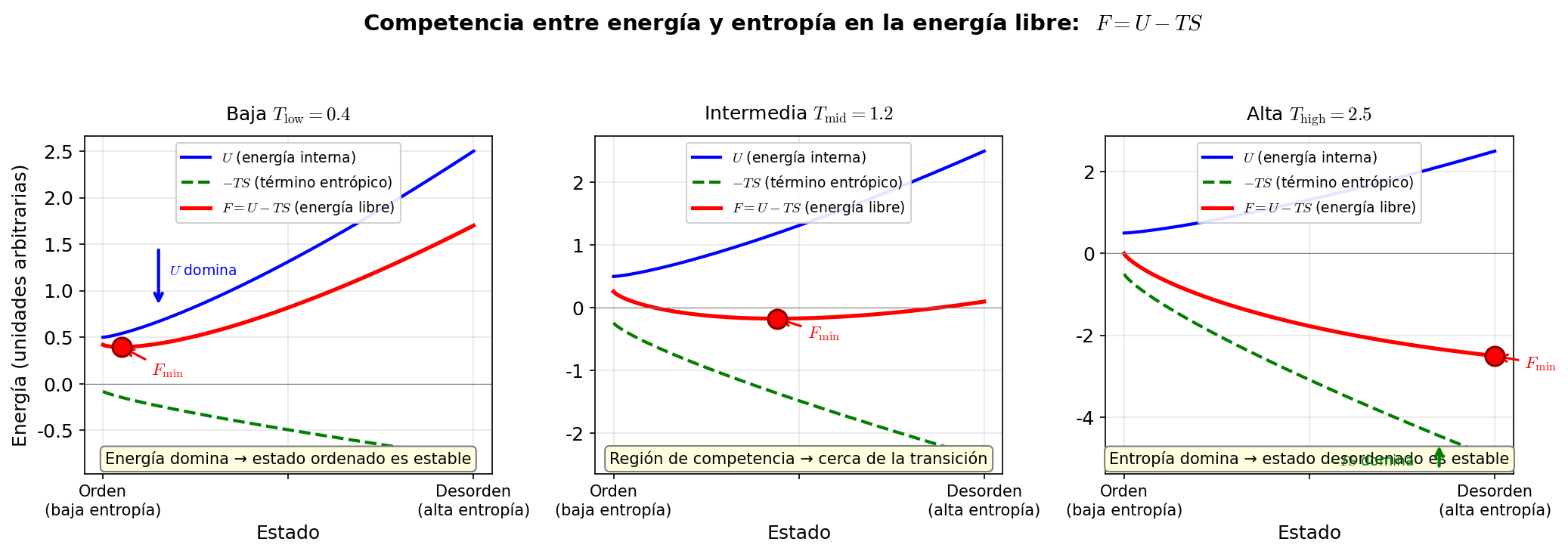
Fig. 3.9: Competencia entre energía y entropía en la energía libre. En \(F = U - TS\), a baja temperatura domina la minimización de energía (orden), y a alta temperatura domina la maximización de entropía (desorden).
🟡 Lina: La esencia de la transición de fase es que la condición de minimización de \(F\) cambia con la temperatura.
Forma diferencial¶
🟡 Lina: Calculemos el cambio infinitesimal de \(F\):
Sustituyendo \(dU = T\,dS - p\,dV\):
🔵 Kai: ¡Oh, \(T\,dS\) se cancela limpiamente!
🟡 Lina: Las variables naturales de \(F\) son \((T, V)\). De las derivadas parciales:
⚪ Mei: Si conoces una sola \(F\), derivando respecto a \(T\) sale la entropía, derivando respecto a \(V\) sale la presión — sale todo.
✅ Verificación de comprensión: ¿Bajo qué condiciones se minimiza la energía libre \(F = U - TS\)?
Respuesta
Bajo condiciones de temperatura constante y volumen constante, el sistema cambia en la dirección que minimiza la energía libre \(F\).
3.9 De la "necesidad" a una "ley fundamental del universo"¶
🟡 Lina: Demos un paso atrás y repasemos la historia de este capítulo.
🔵 Kai: Empezó con la pregunta práctica de querer aumentar la eficiencia de la máquina de vapor.
⚪ Mei: De ahí salió el límite de eficiencia de Carnot, la segunda ley, y se conectó hasta \(S = k_B \ln \Omega\) de Boltzmann.
🔵 Kai: Pero es curioso, ¿verdad? Carnot solo estaba pensando en la eficiencia de la máquina de vapor, ¿por qué llegó a una ley que se aplica a todo el universo?
🟡 Lina: Buena pregunta. Porque Carnot buscó el "límite ideal". En el instante en que descartó los detalles de una máquina concreta y preguntó "¿qué es posible en principio?", el problema trascendió la máquina específica y se volvió universal. La exploración que comenzó con la necesidad práctica de la máquina de vapor alcanzó finalmente una propiedad fundamental del universo: la entropía aumenta. En el prólogo dije que "las motivaciones son de dos tipos: necesidad y curiosidad", pero incluso partiendo de la necesidad se puede llegar a un lugar tan profundo como con la curiosidad.
✅ Verificación de comprensión: ¿De qué pregunta práctica partió la historia de este capítulo, y a qué propiedad fundamental del universo llegó finalmente?
Respuesta
Partió de la pregunta práctica de querer aumentar la eficiencia de las máquinas de vapor, y finalmente alcanzó la propiedad fundamental del universo de que "la entropía aumenta".
3.10 Semilla para el futuro: Entropía y agujeros negros¶
%%{init: {"theme": "default", "themeCSS": ".edgePath .path, .flowchart-link { stroke-width: 2px !important; }"}}%%
flowchart TD
A["Eficiencia de la máquina de vapor<br/>(motivación práctica)"] --> B["Ciclo de Carnot<br/>η = 1 − T_C/T_H"]
B --> C["Segunda ley de la termodinámica<br/>Aumento de la entropía"]
C --> D["Boltzmann<br/>S = k_B ln Ω"]
D --> E["Mecánica estadística<br/>Conteo de microestados"]
E --> F["Bekenstein-Hawking<br/>Entropía de agujero negro<br/>S_BH = A/(4ℓ_P²) k_B"]
F --> G["Strominger-Vafa (1996)<br/>Cálculo de microestados con teoría de cuerdas<br/>(Capítulo 20)"]
style A fill:#ffa,stroke:#333
style G fill:#afa,stroke:#333Fig. 3.10: Genealogía del desarrollo del concepto de entropía
🟡 Lina: Para terminar, voy a dejar una semilla para el futuro.
🔵 Kai: ¿Una semilla?
🟡 Lina: El concepto de entropía que aprendimos en este capítulo reaparecerá en capítulos posteriores. En los años 1970, Bekenstein y Hawking mostraron que los agujeros negros también tienen entropía:
Donde \(A\) es el área del horizonte de sucesos, \(\hbar = h/(2\pi)\) es la constante de Dirac (la constante de Planck \(h\) dividida por \(2\pi\). En mecánica cuántica esta aparece más frecuentemente que \(h\) — por qué lo entenderás en los próximos capítulos. Por ahora piensa que es "una constante de la familia de \(h\)"), y \(\ell_P = \sqrt{G\hbar/c^3}\) es la longitud de Planck (que trataremos en detalle en capítulos posteriores).
🔵 Kai: Los agujeros negros tienen entropía... entonces, pensando desde \(S = k_B \ln \Omega\), ¿los agujeros negros también tienen microestados?
🟡 Lina: Precisamente esa pregunta condujo al mayor éxito de la teoría de cuerdas. En 1996, Strominger y Vafa usaron la teoría de cuerdas para contar los microestados de un agujero negro y derivaron la entropía de Bekenstein-Hawking a partir de \(S = k_B \ln \Omega\) (Cap. 20).
⚪ Mei: ¡Que un concepto nacido de la máquina de vapor se conecte con los agujeros negros!
🟡 Lina: Es lo fascinante de la física, ¿verdad? Campos aparentemente sin relación están conectados en lo profundo. Pero ten cuidado: la entropía de Bekenstein-Hawking surge de la combinación de relatividad general y mecánica cuántica, y su origen microscópico se ha explicado con teoría de cuerdas solo para ciertos agujeros negros (agujeros negros BPS). La extensión a agujeros negros generales es un problema abierto.
🔭 Nota de filosofía de la ciencia: La derivación de la entropía de agujeros negros mediante la teoría de cuerdas es un logro hermoso, pero por sí solo no constituye una "verificación experimental" de la teoría de cuerdas. Porque existe la posibilidad de que el mismo resultado se pueda derivar con otro modelo (como la gravedad cuántica de lazos). "Un modelo que da la respuesta correcta" y "el único modelo correcto" son cosas diferentes. Desde el punto de vista de la falsabilidad, juzga por ti mismo.
✅ Verificación de comprensión: ¿Quiénes mostraron que los agujeros negros también tienen entropía?
Respuesta
Bekenstein y Hawking.
Adelanto del próximo capítulo¶
Cap. 4 — Los tres modelos exitosos —mecánica newtoniana, electromagnetismo y termodinámica— fallan uno tras otro a finales del siglo XIX. La radiación del cuerpo negro, el efecto fotoeléctrico, la precesión del perihelio de Mercurio. Esta "crisis" abre la puerta a las dos grandes revoluciones del siglo XX: la relatividad y la mecánica cuántica. En particular, el problema de la radiación del cuerpo negro desempeña un papel importante como punto de contacto entre la termodinámica aprendida en este capítulo y la teoría cuántica (para más detalles, véase Mecánica Cuántica Cap. 1 en Mecánica Cuántica).
Referencias¶
El contenido de este capítulo se ha estructurado basándose en las siguientes referencias.
- David Tong, Lectures on Statistical Physics, Ch.1: "The Fundamentals of Statistical Mechanics" — Conjunto microcanónico, principio de igual probabilidad a priori
- David Tong, Lectures on Statistical Physics, Ch.2: "Classical Gases" — Entropía de Boltzmann, definición estadística de la temperatura
- David Tong, Lectures on Statistical Physics, Ch.4: "Classical Thermodynamics" — Leyes de la termodinámica, ciclo de Carnot
- David Tong, Lectures on Statistical Physics, Ch.5: "Phase Transitions" — Energía libre
Feedback on this page
Let us know if something was unclear, incorrect, or could be improved.